

Summary
The Intercontinental Lecture, presented by Dr Melvin M. Scheinman, discusses arrhythmogenic right ventricular dysplasia (ARVC). The presenter reviews the history, mechanisms, and pathogenesis of ARVC, with a focus on electrophysiological changes, and then discusses how this new knowledge has modified the clinical approach to the care of this disease.
- antiarrhythmic therapy
- ankyrin-B
- arrhythmias
- arrhythmogenic right ventricular dysplasia/cardiomyopathy
- desmosome protein
- fibroadiposis
- implantable cardioverter defibrillators
- ventricular tachycardia
Melvin M. Scheinman, MD, University of California San Francisco, San Francisco, California, USA, presented the Intercontinental Lecture on arrhythmogenic right ventricular dysplasia/cardiomyopathy (ARVC/ARVC).
ARVC is broadly defined as a structural disease, characterized by replacement of the cardiac myocytes in the free wall of the right ventricle (RV) by fat and/or fibrous tissue. It is associated with ventricular arrhythmias and sudden death. Genetic mutations in desmoplakin, plakoglogin, plakophilin 2, desmoglein 2, and desmocollin 2 are found in 50% to 60% of patients with ARVC [Lombardi R, Marian AJ. PediatrCardiol. 2011]. Some older data implicate the gene RYR2 [Thiene G et al. Orphanet J Rare Dis. 2007], which is also responsible for catecholaminergic polymorphic ventricular tachycardia.
First described over 300 years ago, ARVC is explained as a disruption of cell-to-cell communication, particularly in the desmosomes and more specifically in desmocollin and desmoglein. The desmosomes support structural stability through cell-to-cell adhesion, and regulate transcription of genes involved in adipogenesis and apoptosis, while maintaining proper electrical conductivity through regulation of the gap junctions and calcium homeostasis (Figure 1) [Marcus FI et al. Circulation. 2010].
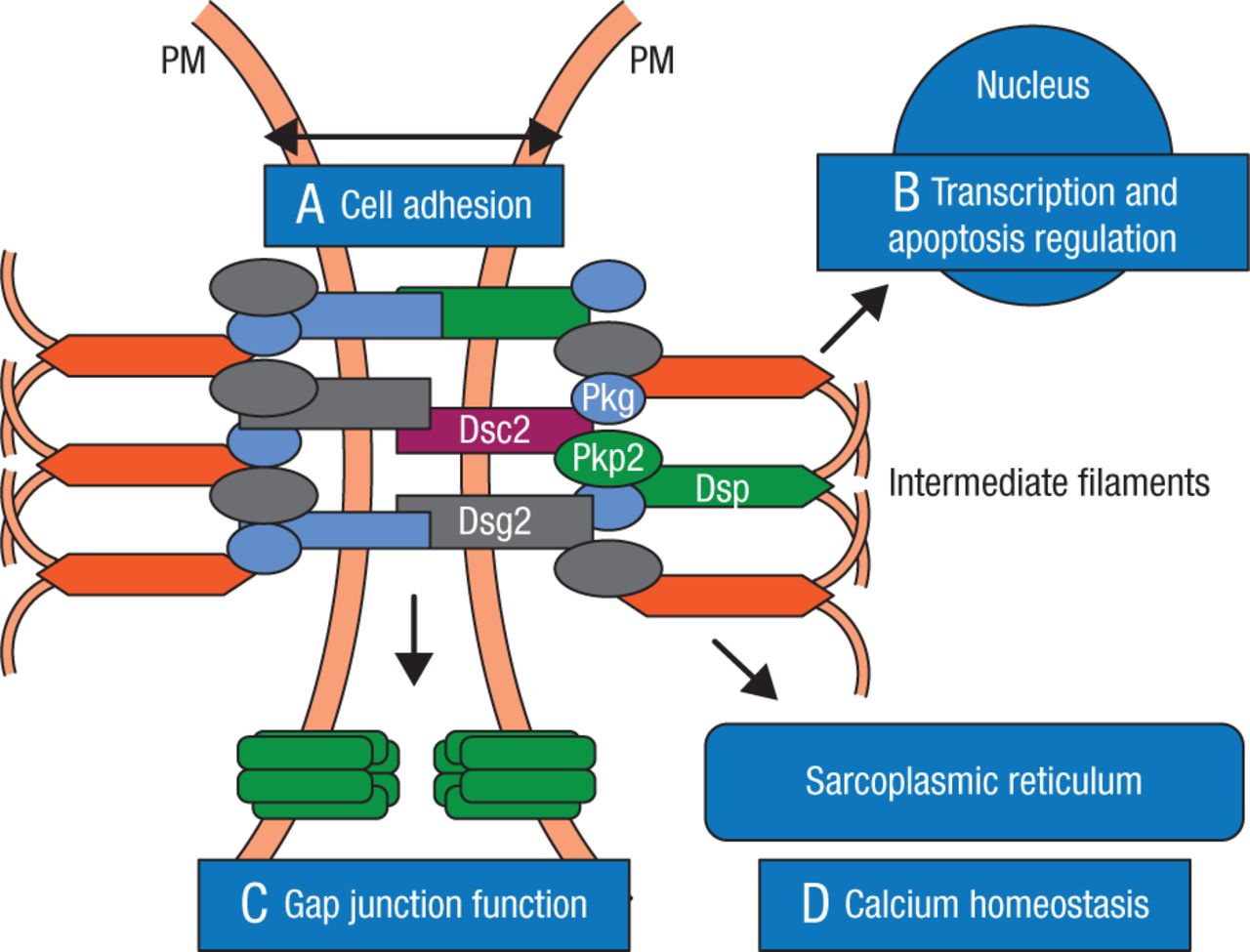
Role of Desmosome in Arrythmogenic Right Ventricular Cardiomyopathy
The cardiac desmosome and proposed roles of the desmosome in (A) supporting structural stability through cell-cell adhesion, (B) regulating transcription of genes involved in adipogenesis and apoptosis, and maintaining proper electrical conductivity through regulation of (C) gap junctions and (D) calcium homeostasis.
Dsc2 indicates desmocollin-2; Dsg2, desmoglein-2; Dsp, desmoplakin; Pkg; plakoglobin; Pkp2, plakophilin-2; and PM, plasma membrane.
Reprinted from Marcus FI et al, Diagnosis of arrhythmogenic right ventricular cardiomyopathy/dysplasia: proposed modification of the task force criteria, Circulation, 2010, Vol 121, Issue 13, Pages 1533–41, with permission from American Heart Association, Inc.
Nuclear translocation of the plakoglogin affects signaling of the nucleus to the cytoplasma, and subsequent suppression of the Wnt signaling system results in a switch to adipogenesis in the second heart-field progenitor cells, a hallmark of the disease ARVC [Lombardi R, Marian AJ. PediatrCardiol. 2011]. Other possible mechanisms may involve a desmosome protein mutation that impairs desmosome assembly, impairing myocyte-to-myocyte attachments and causing cardiac dysfunction, or reduced Ca2+ uptake in the sarcoplasmic reticulum, causing desmosomal disassembly due to elevated cytoplasmic Ca2+ levels and/or impaired Ca2+ homeostasis.
A novel recent finding was the discovery of a connection between the presence of ankyrin-B mutation and exercise that might predispose an individual to ARVC during intense exercise [Roberts JD. Heart Rhythm. 2015]. Echo imaging in such a patient revealed dilated left and right ventricles, mildly reduced left ventricular ejection fraction, and moderate RV enlargement seen on the cardiac magnetic resonance imaging [Roberts JD. Heart Rhythm. 2015]. Genetic abnormalities were noted in ANKB E1458G (a mutation known to cause ankyrin-B syndrome) [Mohler PJ et al. Nature. 2003]. Ankyrins have been implicated in the ARVC phenotype in that they facilitate protein–protein interactions at the voltage-gated sodium channel complex; ankyrin-B has previously been documented to interact with the desmosome [Sato PY et al. Circ Res. 2011].
Dr Scheinman concluded this subsection of his talk by saying that exercise exacerbates the pathophysiology underlying ankyrin-B syndrome, reflective of the pathologic phenotype of ARVC, and should be restricted in patients with the syndrome. ANKB may be a susceptible gene for ARVC and should be included in genetic screening for this disease.
The pathogenesis of ARVC can be summarized as altered integrity of cell-to-cell junctions, connexon function, and sodium channel localization and function. In ARVC, there is an interference with the signaling pathways responsible for normal development of RV muscle. In addition, abnormalities in cytosolic calcium may interfere with normal desmosome structure.
Dr Scheinman turned to recent research that has uncovered a new biomarker and desmoplakin-interacting protein, signalosome subunit 6 (CSN6), which is involved in the regulation of protein degradation and can increase the accuracy of predicting the diagnosis of ARVC. In the clinic, Dr Scheinman and colleagues are looking at the long-term changes in electrical patterns in ARVC patients such as T-wave inversions, epsilon waves, QRS fragmentation, and terminal activation delay. They have found that precordial lead T-wave inversion was more common at baseline and had the highest progression in ARVC patients, reflecting involvement of subpulmonic valve and RV outflow tract. Inferior lead abnormalities were the second most common at baseline and were considered an early marker for disease progression.
Looking at the changes in the echocardiographic findings to determine RV structural and functional abnormalities, the only significant finding was a mean annual increase in the RV end-diastolic area of 2.9%, far greater than any other RV structural change. However, there was no correlation between the electrical changes and the RV structure and functional changes. Radionuclear measurements show high sensitivity (81% sensitivity) for detecting ARVC [Svetlichnaya J. Heart Rhythm. 2015], better than echocardiography or magnetic resonance imaging [Johnson CJ. Heart Rhythm. 2015]
β-blockers (sotalol), amiodarone, catheter ablation, and defibrillators have also been used to treat ARVC with some success. In an early study, sotalol at high doses (320 to 480 mg/d) proved to be highly effective in ARVC patients with inducible as well as noninducible ventricular tachycardia compared with other classes of agents such as sodium channel blockers, β-blockers, amiodarone, verapamil, and a combination of agents [Wichter et al. Fighting Sudden Cardiac Death: A Worldwide Challenge. 2000].
In a more recent study in 95 North American registry patients, low-dose sotalol (160 to 320 mg/d) was reported to be equivalent to β-blockers, whereas amiodarone proved most effective in preventing ventricular arrhythmias for treating patients with ARVC [Marcus CA et al. J Am Col Cardiol. 2009]. In patients who were refractory to single-drug therapy and ablation, the arrhythmias were ultimately controlled with combinations of sotalol, flecainide, and amiondarone [Ermakov S et al. Pacing Clin Electrophysiol. 2014]. Appropriate implantable cardioverter defibrillator interventions are also recommended for primary prevention among patients with ARVC. In a meta-analysis, annualized cardiac mortality and heart transplant rates were both 0.9% in patients with ARVC who had an ICD for primary or secondary prevention of sudden cardiac death, although the annualized inappropriate implantable cardioverter defibrillator intervention rate was relatively high (3.7%) [Schinkel AF. Circ Arrhythm Electrophysiol. 2013]. Complications included difficult lead placement (18.4%), lead malfunction (9.8%), and displacement (1.4%). Early results in 2 patients from a study using decongestant therapy (combination of isosorbide dinitrate and spironolactone/hydrochlorothiazide) to treat ARVC showed an improvement or lack of progression of RV size and function, and a decrease in ventricular tachycardia/ventricular fibrillation burden after 12 months of therapy. This study will be expanded to include echocardiograms looking at more sensitive parameters during exercise at baseline and after 12 months of therapy. Newer techniques involving cardiac biopsies as well as the use of induced pluripotent stem cells show great promise in better defining mechanisms.
- © 2015 SAGE Publications
Tools









{kind=link}
Table of contents
Cited By...
- No citing articles found.