

Summary
This article discusses the pathophysiology of osteogenesis imperfecta (OI), in particular in relation to some rare, recessive forms of the condition. Although historically considered a collagen-related condition, predominantly due to mutations in genes that encode the alpha chains of collagen type I, mutations in various noncollagenous genes have also more recently been discovered to cause some forms of OI.
- Metabolic Bone Disease
- Metabolic Bone Disease
- Endocrinology
- Diabetes & Metabolic Syndrome
Frank Rauch, MD, Shriners Hospital for Children, Montreal, Quebec, Canada, discussed the pathophysiology of osteogenesis imperfecta (OI), in particular in relation to some rare, recessive forms of the condition. Although historically considered a collagen-related condition, predominantly due to mutations in genes that encode the alpha chains of collagen type I, mutations in various noncollagenous genes have also more recently been discovered to cause some forms of OI.
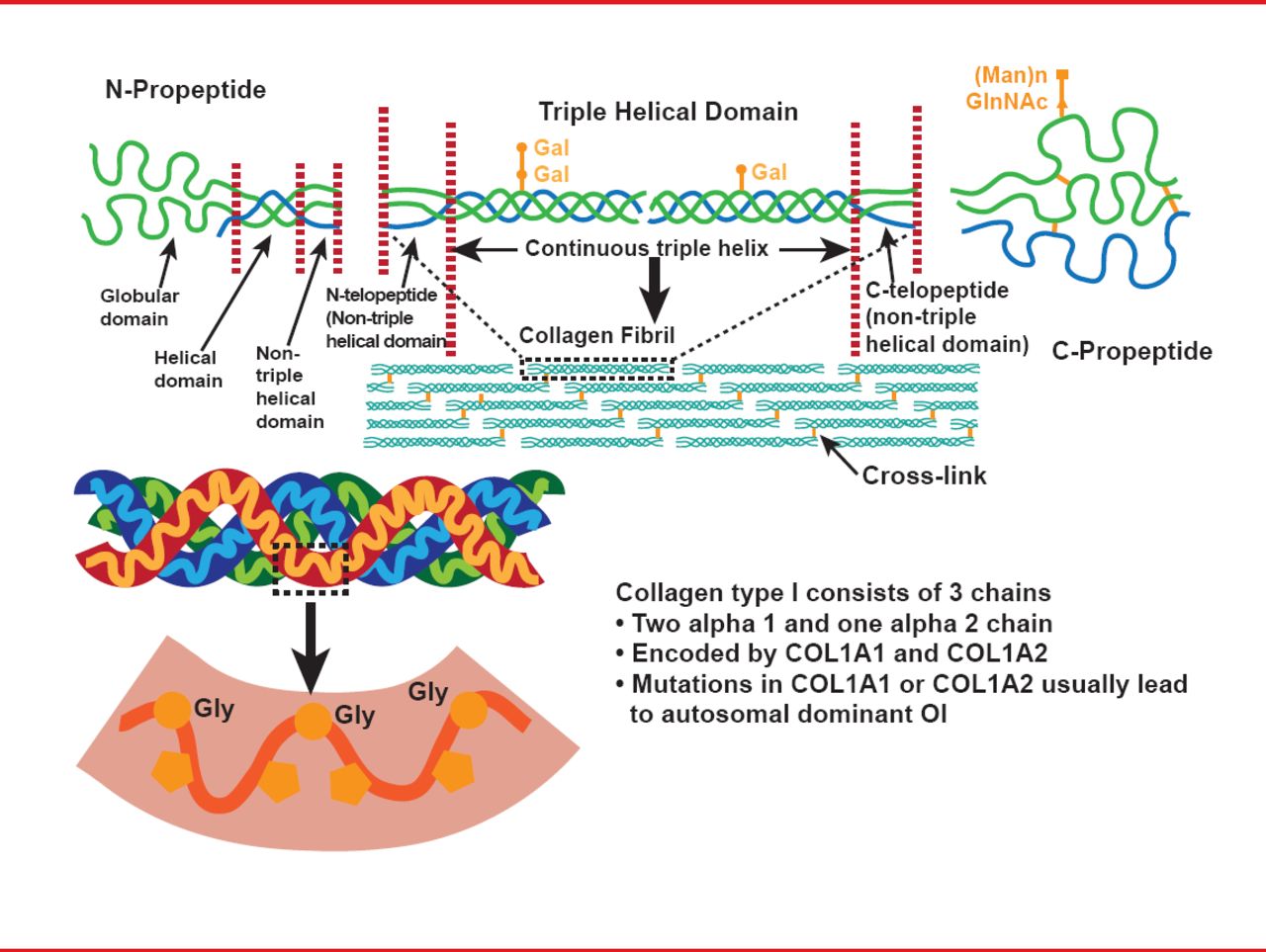
This inherited bone disorder is typically characterized by reduced bone mass, bone fragility, and often short stature. Most cases are due to dominant mutations in one of the two genes that encode alpha chains of collagen type I (COL1A1 or COL1A2; Figure 1), and disease severity varies across a range of four classical phenotypes: type I is the mildest form, characterized by patients with straight legs and spine; type II is the perinatal lethal form; type III is the most severe form seen in survivors; and type IV is intermediate in severity between types I and III. Some extraskeletal manifestations are also variably associated, including blue sclera, dentinogenesis imperfecta, and joint hyperlaxity [Sillence DO et al. J Med Genet 1979].

The Structure of Type I Collagen
Reproduced with permission from F Rauch, MD.
However, recessive types of OI have now also been described, caused by mutations in noncollagenous genes involved in various aspects of bone formation. OI due to mutations in genes associated with proteins involved in collagen type I processing are rare, but 8 forms are currently known. These include mutations in prolyl 3-hydroxylase 1, a gene that codes for an enzyme involved in collagen alpha 1 chain synthesis [Cabral WA et al. Nat Genet 2007]; FKBP65, which codes for a protein involved in formation of a triple helix from the alpha chains; and BMP1, which codes for an enzyme responsible for cleavage of the alpha chain [Marini JC, Blissett AR. J Clin Endocrinol Metab 2013].
In contrast to these types with autosomal recessive inheritance, OI type V is caused by a dominant defect in IFITM5, the gene that encodes BRIL protein [Marini JC, Blissett AR. J Clin Endocrinol Metab 2013]. Phenotypically, this condition is associated with striking development of hyperplastic callus [Glorieux FH et al. J Bone Miner Res 2000], but although all patients have the same point mutation in the 5'-UTR of the gene, how this leads to this OI phenotype and bone fragility remains unknown [Rauch F et al. J Med Genet 2013; Cho TJ et al. Am J Hum Genet 2012; Semler Oet al. Am J Hum Genet 2012].
In addition to these forms of OI that are linked to collagen processing in some way, other gene defects have been described, although their exact association with collagen is unknown.
The gene SERPINF-1 codes for a protein known as pigment-epithelium derived factor (PEDF), and mutations in this gene produce a disease phenotype that was originally characterized as OI type VI. Clinically, children with this type are born without bone deformities, and do not experience their first fracture until they are 6 to 12 months old, but then develop progressively more fractures, leading to bone deformity [Homan EP et al. J Bone Miner Res 2011].
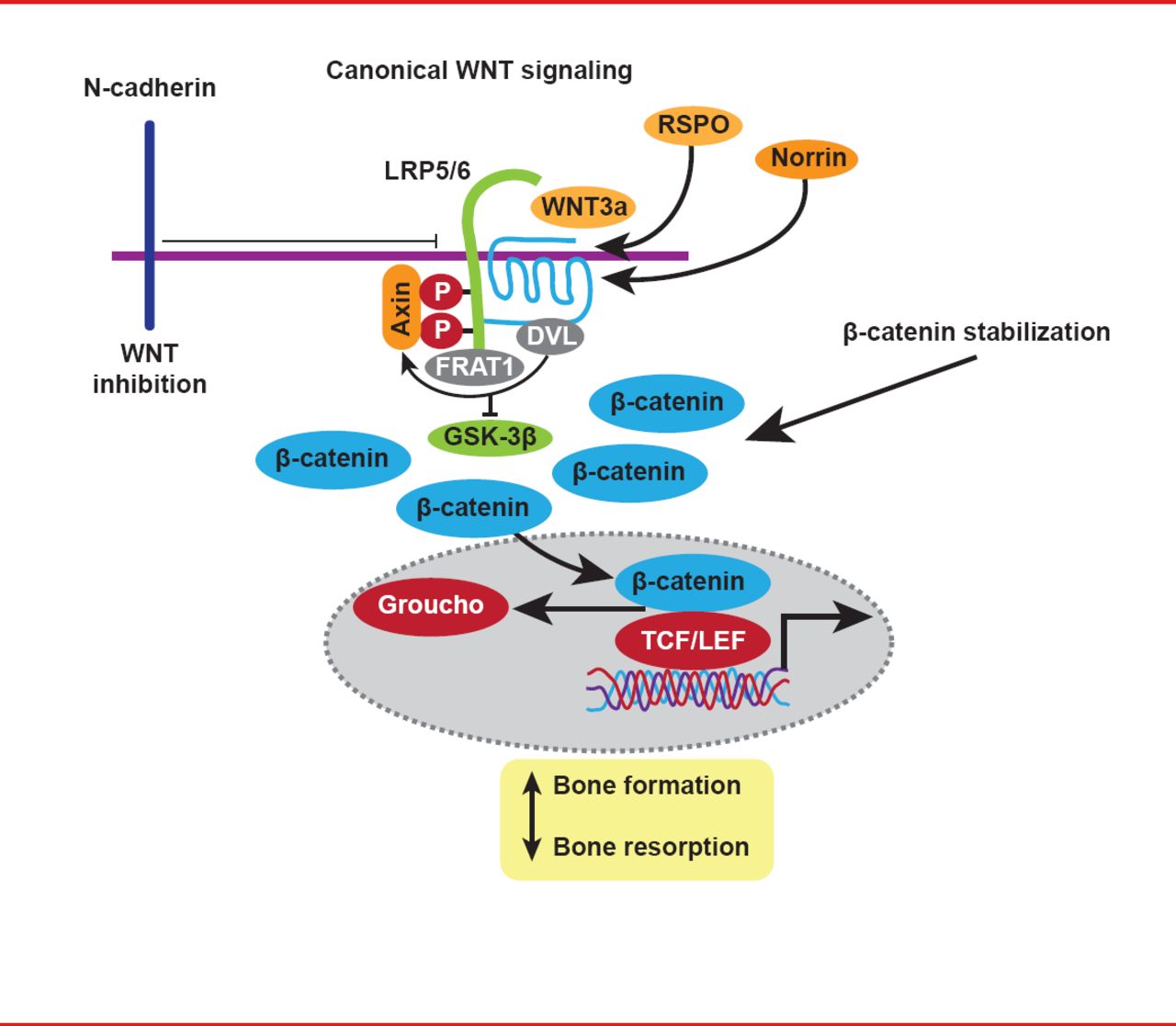
WNT proteins and their signaling cascades are key regulators of osteoblast activity, and mutations in the gene WNT1 have also been shown to cause OI, due to loss of WNT signaling (Figure 2) [Fahiminiya S et al. J Med Genet 2013].

The WNT Signaling Pathway
Reproduced with permission from F Rauch, MD.
Despite the paradigm shift for OI as a collagen-associated disorder, Prof. Rauch concluded by raising some unsolved philosophical questions relating to the more recent genetic findings, specifically as to whether OI should be defined by its clinical picture or in relation to its affected genes.
- © 2013 MD Conference Express®
Tools









{kind=link}
{kind=link}
Table of contents
Cited By...
- No citing articles found.