

Summary
Idiopathic pulmonary fibrosis (IPF) is a distinctive form of chronic interstitial lung disease occurring primarily in older adults, more often male [Raghu G et al. Am J Respir Crit Care Med 2011]. The estimated prevalence of IPF is 14 to 43 per 100,000 and an estimated incidence is 7 to 16 per 100,000, depending on how it is defined [Raghu G et al. Am J Respir Crit Care Med 2006]. Prognosis can be predicted by the radiographic extent of the disease, amount of fibrosis, presence of pulmonary hypertension, and comorbidities (emphysema, lung cancer, and coronary artery disease).
- Cancer
- Coronary Artery Disease
- Thromboembolic Disease Lower Respiratory Infections
- Pneumonia
- Oncology
- Pulmonary & Respiratory Medicine
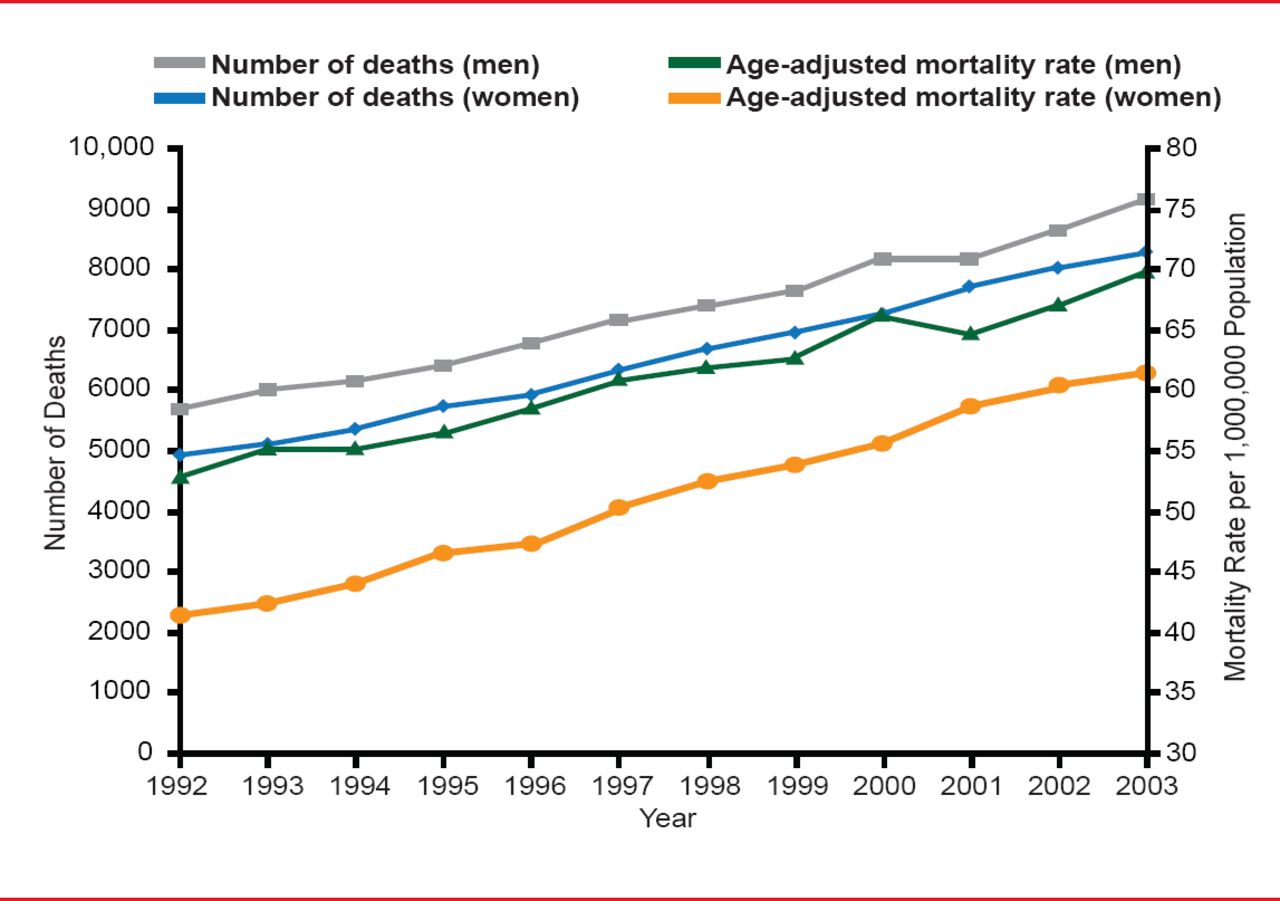
Gregory Tino, MD, University of Pennsylvania School of Medicine, Philadelphia, Pennsylvania, USA, defined idiopathic pulmonary fibrosis (IPF) as a distinctive form of chronic interstitial lung disease occurring primarily in older adults, more often male [Raghu G et al. Am J Respir Crit Care Med 2011]. The estimated prevalence of IPF is 14 to 43 per 100,000 and an estimated incidence is 7 to 16 per 100,000, depending on how it is defined [Raghu G et al. Am J Respir Crit Care Med 2006]. IPF is often associated with smoking, environmental factors, viral factors, and chronic aspiration associated with gastroesophageal reflux disease (GERD), although its etiology is not well understood. Mortality is high, increasing, and most often due to pulmonary fibrosis, cardiovascular disease, lung cancer, or pneumonia (Figure 1). Prognosis can be predicted by the radiographic extent of the disease, amount of fibrosis, presence of pulmonary hypertension, and comorbidities (emphysema, lung cancer, and coronary artery disease [CAD]). The median time to death after diagnoses is 3.2 years [Ley B et al. Ann Intern Med 2012], based on the GAP (Gender, Age and Physiology) staging system, a simple scoring model that uses commonly measured clinical and physiologic variables to predict mortality in patients with IPF.

IPF Epidemiology: Mortality Rates
Reprinted with permission of the American Thoracic Society. Olson AL et al. Mortality from Pulmonary Fibrosis Increased in the United States from 1992 to 2003. Am J Respir Crit Care Med 2006;176:277–284. Official journal of the American Thoracic Society.
Acute exacerbations of IPF are common and frequently fatal, but not well understood. Harold R. Collard, MD, University of California, San Francisco, San Francisco, California, USA, reported that acute exacerbations of IPF represent a distinct entity defined as acute, clinically significant deteriorations of unidentifiable cause in patients with underlying IPF. Proposed diagnostic criteria include subjective worsening over ≤30 days, new bilateral radiographic opacities, the absence of microbiological evidence of infection, and exclusion of other causes of acute worsening [Collard HR et al. Am J Respir Crit Care Med 2007]. Acute exacerbation is a significant predictor of poor survival, and appears to be more common in patients with low forced vital capacity and diffusing capacity of the lung for carbon monoxide [Song JW et al. Eur Respir J 2011].
Potential triggers of acute exacerbation are viral infection, surgical procedures, microaspiration, and ambient pollution. Preventive measures, including vaccination, avoidance of unnecessary surgery and pollutant exposures, and evaluation and management of GERD, may have a role. Treatment is largely supportive but most patients with acute exacerbation will be treated with antibiotics and corticosteroids [Collard HC et al. Resp Med 2007]. There is a >50% mortality at 90 days in most cohorts of patients post-acute exacerbation with a median survival of 2.2 months [Song JW et al. Eur Respir J 2011].
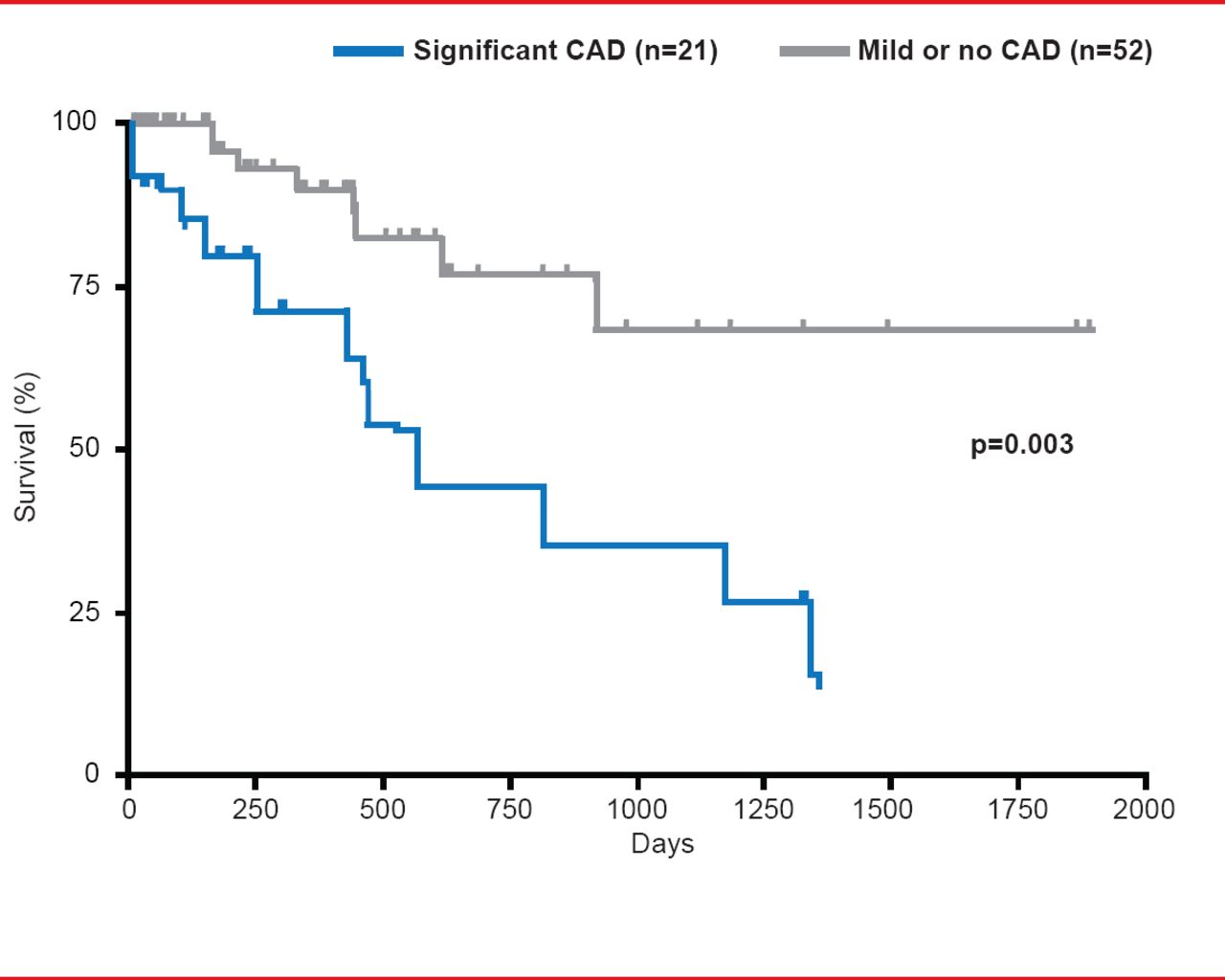
CAD has a high prevalence in patients with IPF and has a significant impact on mortality (Figure 2). In a recent study, patients with IPF were more significantly likely to also have CAD compared with chronic obstructive pulmonary disease (COPD; 65.8% vs 46.1%; p<0.028). Unsuspected significant CAD was found in 18% of IPF patients versus 10.9% of those with COPD (p<0.004) [Nathan SD et al. Respir Med 2010]. In his presentation Steven D. Nathan, MD, George Mason University, Virginia Commonwealth University, Falls Church, Virginia, USA, speculated that IPF might promote atherosclerosis through either cytokine cross talk or plaque instability leading to hypoxia. Although not significant, IPF status appeared predictive of the presence of CAD (adjusted OR, 1.67; 95% CI, 0.59 to 4.78) [Nathan SD et al. Respir Med 2010] as well as for the occurrence of first-time coronary syndromes [Hubbard RB et al. Am J Respir Crit Care Med 2008].
Dr. Nathan stated that coronary calcification, assessed by routine CT of the chest, is a good technique for predicting underlying significant CAD in patients with IPF. The sensitivity of moderate to severe calcification was 81%, while the specificity was 85%, with an associated OR of 25.2 (4.64 to 166, p <0.005) with excellent radiologist and pulmonologist agreement in the grading of the coronary calcification [Nathan SD et al. Respirology 2011; Weir N et al. Am J Respir Crit Care Med 2010].

Influence of CAD on IPF Mortality
Reproduced from Nathan SD et al. Prevalence and impact of coronary artery disease in idiopathic pulmonary fibrosis. Respir Med 2010;104(7):1035–1041.
Mary Elizabeth Kreider, MD, Hospital of the University of Pennsylvania, Philadelphia, Pennsylvania, USA, discussed the concomitance of IPF and lung cancer, the latter, of which appears elevated in IPF patients. The frequency of lung cancer in patients with IPF and varies widely by study type and country with prevalence rates of 3% to 48% being reported compared with a non-IPF population. Among more recent studies the lung cancer prevalence rate is in the 5% to 10% range. Risk factors may include male gender, increasing age, and smoking history. Cancer cells are more likely to be peripheral and in lower lobes but not all are near fibrotic areas. Dr. Kreider suggested three possibilities for the link between lung cancer and IPF: IPF causes lung cancer, lung cancer causes IPF, or lung cancer and IPF occur independently but share a common pathogenic mechanism. The impact of the presence of cancer in IPF patients on survival is not clear but survival from time of dual diagnosis is likely poor. Therapy is complicated by high rates of acute exacerbation and difficulty in tolerating interventions.
When a clot from a venous thromboembolism (VTE) comes in contact with IPF, there is an increased risk of worse outcome compared with either condition alone. Amy L. Olson, MD, MSPH, University of Colorado, Denver, Colorado, USA, addressed the question of whether the coagulation pathway is activated by IPF or VTE triggers IPF.
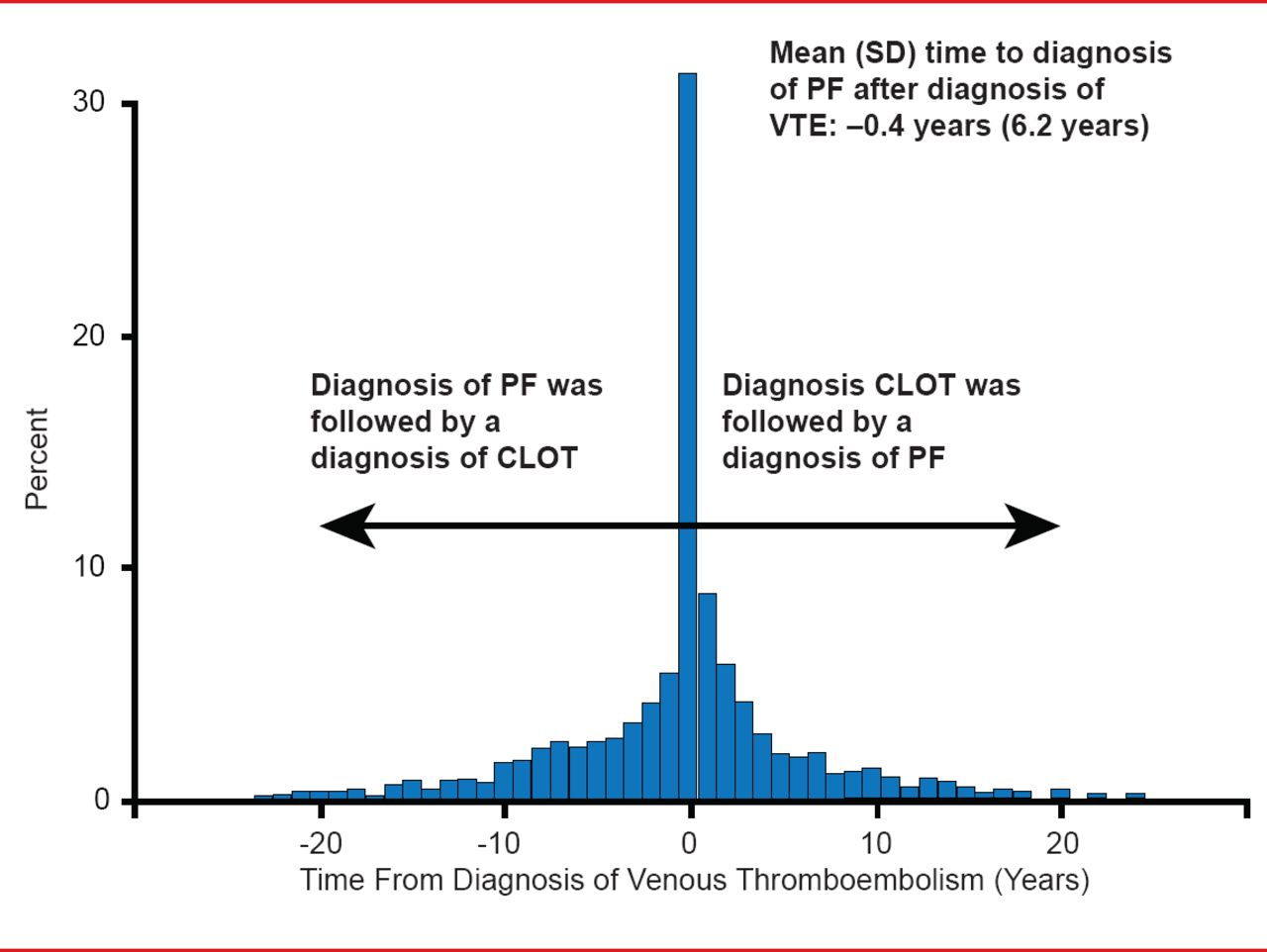
Although not found in the normal lung, tissue factor (TF) and fibrin have been found in the pneumocytes of IPF patients [Imokawa S et al. Am J Respir Crit Care Med 1997]. In addition, TF levels are significantly higher in advanced versus nonadvanced cases, indicating that excessive procoagulant activity in the alveolar space may play a relevant role in the pathogenesis of IPF [Fuji M et al. Thromb Res 2000]. If this hypothesis is true, IPF patients are at increased risk of vascular disease compared with the general population. This effect is most marked for acute coronary syndrome and deep-vein thrombosis (DVT), which is diagnosed at a rate of 5.9/1000 person years in patients with IPF versus 2.1/1000 person years in the general population [Hubbard RB et al. Am J Respir Crit Care Med 2008]. Time from a VTE event to a diagnosis of IPF is −0.4 years. A diagnosis of IPF is just as likely to follow a diagnosis of VTE as to precede it (Figure 3) [Sode BF et al. Am J Respir Crit Care Med 2010].

Pulmonary Fibrois and VTE
Reprinted with permission of the American Thoracic Society. Sode BF et al. Venous Thromboembolism and Risk of Idiopathic Interstitial Pneumonia: A Nationwide Study. Am J Respir Crit Care Med. 2010;181:1085–1092. Official journal of the American Thoracic Society.
The presence of VTE can result in a greater risk of pulmonary fibrosis, COPD, and lung cancer, the combination of which leads to earlier deaths in both men (72.0 vs 74.4 years) and women (74.3 vs 77.4 years; p<0.0001 for both) [Sprunger DB et al. Eur Respir J 2012].
Pulmonary hypertension (PH) is hemodynamic and pathophysiological condition characterized by the pulmonary arterial pressure ≥25 mm Hg [Galie N et al. Eur Respir J 2009]. Precapillary PH is common in patients with severe IPF and is associated with increased mortality and a worse clinical course. MJC Humbert, MD, PhD, Hôpital Antoine Béclère, Université Paris-Sud, Clamart, France, commented that PH usually is of mild-to-moderate severity (mPAP 25 to 35 mm Hg), with slow progression and preserved right ventricular function. Severe PH (mPAP >35 mm Hg) may be found in a minority of patients, with some similarities with idiopathic PAH (in this subgroup. Therapies used in PH are generally discouraged (poor efficacy, possible worsening of gas exchange); however, long-term oxygen therapy is the most often therapy of choice if one is used. More randomized controlled trials are needed to test newer treatments.
- © 2013 MD Conference Express®
Tools









{kind=link}
{kind=link}
{kind=link}
Table of contents
Cited By...
- No citing articles found.