

Summary
Atherosclerosis is caused by the combined effect of lipid infiltration and inflammatory cytokine activation in large and medium arterial walls. A direct correlation exists between coronary heart disease, vascular disease, and atherosclerosis with risk thresholds starting at very low or normal risk factor levels. This article discusses the angiotensin receptor blockade, as well how to target the renin-angiotensin-aldosterone system for atherosclerotic vascular disease, vascular remodeling, and hypertension.
- Hypertensive Disease
- Coronary Artery Disease Lipid Disorders
Angiotensin Receptor Blockade and Atherosclerotic Disease
Atherosclerosis is caused by the combined effect of lipid infiltration and inflammatory cytokine activation in large and medium arterial walls. A direct correlation exists between coronary heart disease, vascular disease, and atherosclerosis with risk thresholds starting at very low or normal risk factor levels: blood pressure (BP; 110/70 mm Hg), low-density lipoprotein cholesterol (LDL-C; 70 mg/dL), and serum glucose (75 mg/dL fasting blood glucose). Traditionally, physicians diagnosed and treated cardiovascular (CV) risk factors independently, but it is now understood that risk factors often occur concurrently, producing a multiplicative effect on CV risk.
Angiotensin II plays a central role in the progression of vascular disease from hypertension to atherosclerosis to myocardial infarction, vascular and myocardial remodeling, and heart failure, culminating in end-organ failure and death. Agents that interrupt the renin-angiotensin-aldosterone system (RAAS) lower BP and improve vascular structural and functional changes associated with hypertension. Carlos M. Ferrario, MD, Wake Forest University School of Medicine, Winston-Salem, North Carolina, USA, discussed the effects of angiotensin receptor blockade on the progression of atherosclerosis.
BP is regulated by the interactions of the RAAS, volume status, and the sympathetic nervous system (SNS). The RAAS regulates sodium and volume balance, affecting vascular tone and SNS activity through the following process: renin released from the kidneys catalyzes the conversion of angiotensinogen to angiotensin I, which is converted to angiotensin II by angiotensin-converting enzyme (ACE). Circulating angiotensin II also stimulates aldosterone synthesis, leading to sodium and water reabsorption, which increases plasma volume, total peripheral resistance, and BP.
Angiotensin II binds to angiotensin II type I receptors (AT1R), leading to a series of steps that are thought to ultimately result in mitogen-activated protein kinase activation, which mediates angiotensin II induced cell proliferation and apoptosis. Evidence suggests that angiotensin II is involved in restructuring of the arterial wall in both atherosclerosis and hypertension. Angiotensin II promotes monocyte recruitment into the vascular intima and indirectly facilitates macrophage and smooth muscle cell transformation into foam cells by promoting superoxide radical formation. This oxidative stress enhances LDL oxidation and nitric oxide degradation. AT1R crosstalk with the oxidized LDL receptor (LOX-1) promotes the incorporation of oxidase LDL into macrophages and their consequent transformation into foam cells.
Targeting the RAAS: Atherosclerotic Vascular Disease
Numerous large clinical trials have studied the effects of ACE inhibition on CV disease, but the effects of angiotensin receptor blockade on hypertensive atherosclerotic vascular disease are poorly understood. In a primate model of atherosclerosis, the angiotensin receptor blocker (ARB) losartan significantly reduced atherosclerosis in the coronary arteries (p<0.01) [Strawn WB et al. Circulation 2000]. A study in patients with coronary artery disease showed that angiotensin II AT1R blockade reduces monocyte CD11b expression [Khan BV et al. J Am Coll Cardiol 2001].
The LIFE trial, a landmark study in 2002 [Dahlof B et al. Lancet 2002], reported that an ARB versus an atenolol-based therapy significantly reduced the risk of stroke (RR, 0.79; 95% CI, 0.69 to 0.90), HF (RR, 0.84; 95% CI, 0.72 to 0.97), and major CV events (RR, 0.90; 95% CI, 0.83 to 0.96) in patients aged 55 to 80 years with essential hypertension and left ventricular hypertrophy.
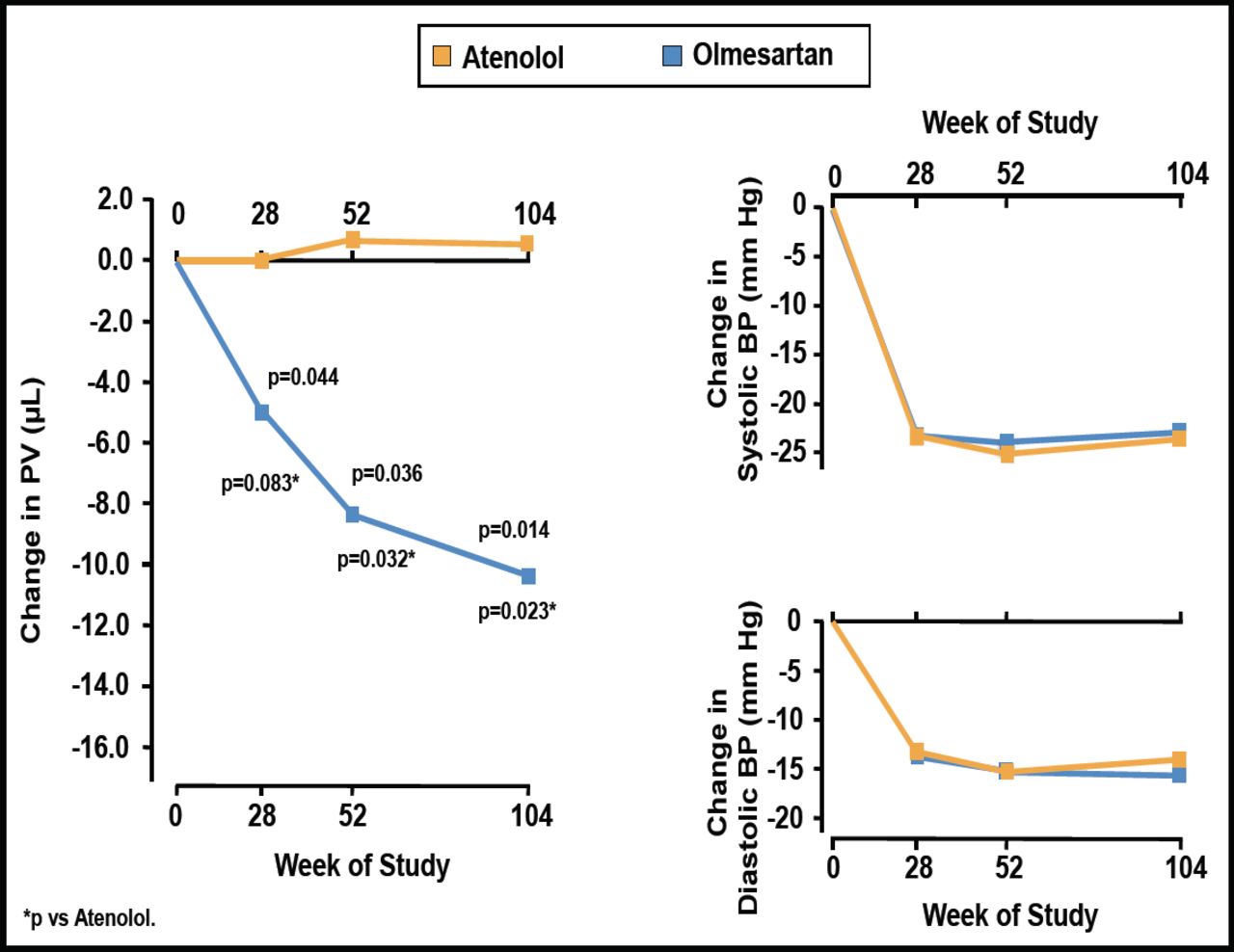
Stumpe et al. [Ther Adv Cardiovasc Dis 2007] found that the ARB olmesartan significantly reduced plaque volume from baseline at 28 (p=0.044), 52 (p=0.036), and 104 (p=0.014) weeks of follow-up, while atenolol had no effect on plaque volume (Figure 1). Olmesartan and atenolol caused similar reductions in BP.

Changes from Baseline in Plaque Volume and BP with Olmesartan versus Atenolol.
BP=blood pressure; PV=plaque volume;
Reprinted from Ther Adv Cardiovasc Dis 2007;1(2):97. Stumpe KO et al. The Multicenter Olmesartan Atherosclerosis Regression Evaluation (MORE) Study, with permission from Sage Journals.
Hirohata et al. [J Am Coll Cardiol 2010] randomized 247 patients with stable angina to olmesartan versus control to investigate the effect of angiotensin II blockade on atherosclerosis progression. Serial intravascular ultrasound examinations at baseline and 14 months showed that the change in nominal atheroma volume was significantly greater with olmesartan (−2.6 mm3; 95% CI, −7.9 to 2.8) versus control (7.1 mm3; 95% CI, 1.8 to 12.4; p=0.011).
Targeting the RAAS: Vascular Remodeling and Hypertension
Small resistance vessel remodeling may be the earliest manifestation of hypertension. Angiotensin II has a prominent role in vascular remodeling, which may be the primary cause of the development of hypertension. Schiffrin et al. [Circulation 2000] demonstrated that losartan versus atenolol reversed vascular hypertrophy independent of BP after 1 year of treatment (p<0.05).
Prehypertension is a strong predictor of CV risk. The Trial of Preventing Hypertension [TROPHY] study investigated whether early treatment of high-normal BP with candesartan (n=391) versus placebo (n=381) would prevent or postpone development of stage 1 hypertension [Julius S et al. N Engl J Med 2006]. After 4 years, hypertension had developed in 240 subjects in the placebo group and 208 subjects in the candesartan group (candesartan RR reduction, 15.6%; p<0.007).
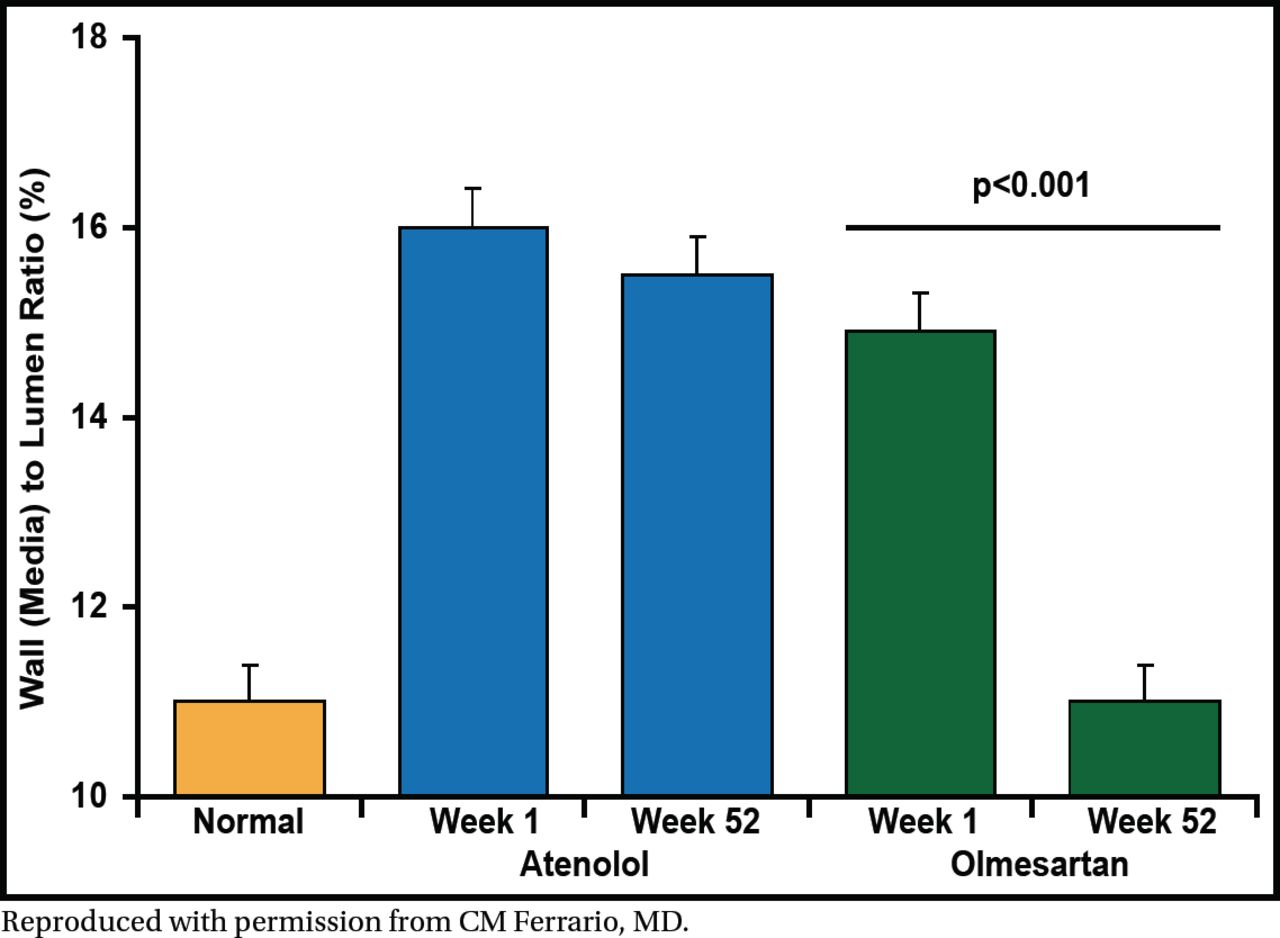
In the Vascular Improvement with Olmesartan Study [VIOS], Dr. Ferrario's group investigated the changes in small subcutaneous resistance arterioles in stage I non-diabetic hypertensive patients (n=100, 61% male, aged 38 to 67 years) randomized to olmesartan medoxomil (20 to 40 mg) versus atenolol (50 to 100 mg) plus hydrochlorothiazide, amlodipine, or hydralazine as needed [Smith RD et al. J Am Soc Hypertension 2008]. The primary endpoint was BP ≤140/90 mm Hg. At baseline and after 1 year of treatment, subcutaneous gluteal resistance arteries were examined on a pressurized myograph to evaluate remodeling.
After 52 weeks of treatment, the target BP was achieved in both treatment arms. Vascular hypertrophy was reversed from baseline to week 52 with olmesartan treatment (p<0.001) but not with atenolol (Figure 2). Central aortic pressure and augmentation index were significantly reduced from baseline to 52 weeks in the olmesartan arm (p<0.05 for both) but not in the atenolol arm. These results show that reversal of vascular hypertrophy is mediated by blockade of the AT1-dependent actions of angiotensin II.

Olmesartan Medoxomil but Not Atenolol Reverses Vascular Hypertrophy.
Reproduced with permission from CM Ferrario, MD.
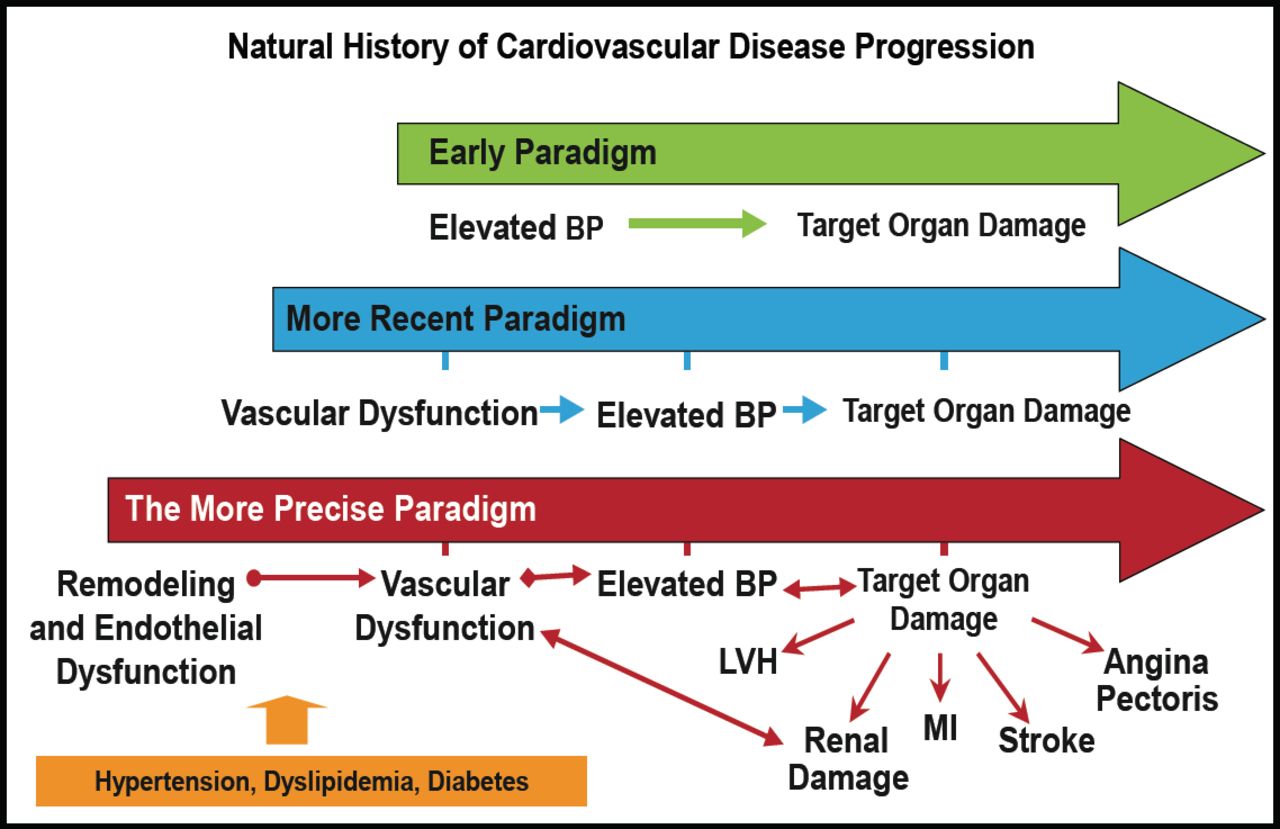
According to Dr. Ferrario, these studies of angiotensin receptor blockade have resulted in a more precise paradigm for the development of vascular disease and its consequences. Hypertension, dyslipidemia, and diabetes lead to vascular remodeling and endothelial dysfunction, resulting in vascular dysfunction that causes further elevations in BP, and ultimately results in target organ damage (Figure 3).

The Continuum of Vascular Disease.
BP=blood pressure; CVD=cardiovascular disease; LVH=left ventricular hypertrophy; MI=myocardial infarction.
Reproduced with permission from CM Ferrario, MD.
- © 2012 MD Conference Express®
Tools









{kind=link}
{kind=link}
{kind=link}
Table of contents
Cited By...
- No citing articles found.