

Summary
The Acute Study of Clinical Effectiveness of Nesiritide in Decompensated Heart Failure [ASCEND-HF; NCT00475852] was designed as the result of an independent safety and efficacy review of nesiritide data in an attempt to assess the concerns that were raised by the 2005 reports more fully.
- Cardiology Clinical Trials
- Heart Failure
Results from a large, prospective clinical trial that was designed to assess the safety and efficacy of nesiritide that was added to standard care in patients with acute decompensated heart failure (ADHF) showed that nesiritide is safe but offers no significant benefit in terms of mortality or HF rehospitalization rates. There was a modest improvement in dyspnea. Renal function was not compromised.
Nesiritide is a recombinant intravenous (IV) formulation of human B-type natriuretic peptide that is known to reduce dyspnea and intracardiac filling pressures within 3 hours of administration in patients with ADHF. It was approved in 2001 to reduce pulmonary capillary wedge pressure and improve dyspnea and was widely used until 2005, when the results of two meta-analyses questioned its safety, noting a higher mortality rate [Sackner-Bernstein JD et al. JAMA 2005] and increased risk of kidney injury [Sackner-Bernstein JD et al. Circulation 2005]. The Acute Study of Clinical Effectiveness of Nesiritide in Decompensated Heart Failure (ASCEND-HF; NCT00475852) was designed as the result of an independent safety and efficacy review of nesiritide data in an attempt to assess the concerns that were raised by the 2005 reports more fully.
ASCEND-HF was a prospective, double-blind, randomized trial in 7141 patients (median age 67 years; ∼34% women) with ADHF, dyspnea at rest or with minimal activity, and one clinical sign and one objective measure of HF. Within 24 hours of hospitalization, subjects were randomly assigned to receive either IV nesiritide (n=3496; initial IV bolus of 2 μg/kg at the discretion of the investigator, followed by continuous IV infusion of 0.01 μg/kg) or matching placebo (n=3511) for up to 7 days, along with usual care. The duration of treatment was based on the investigator's assessment of clinical improvement.
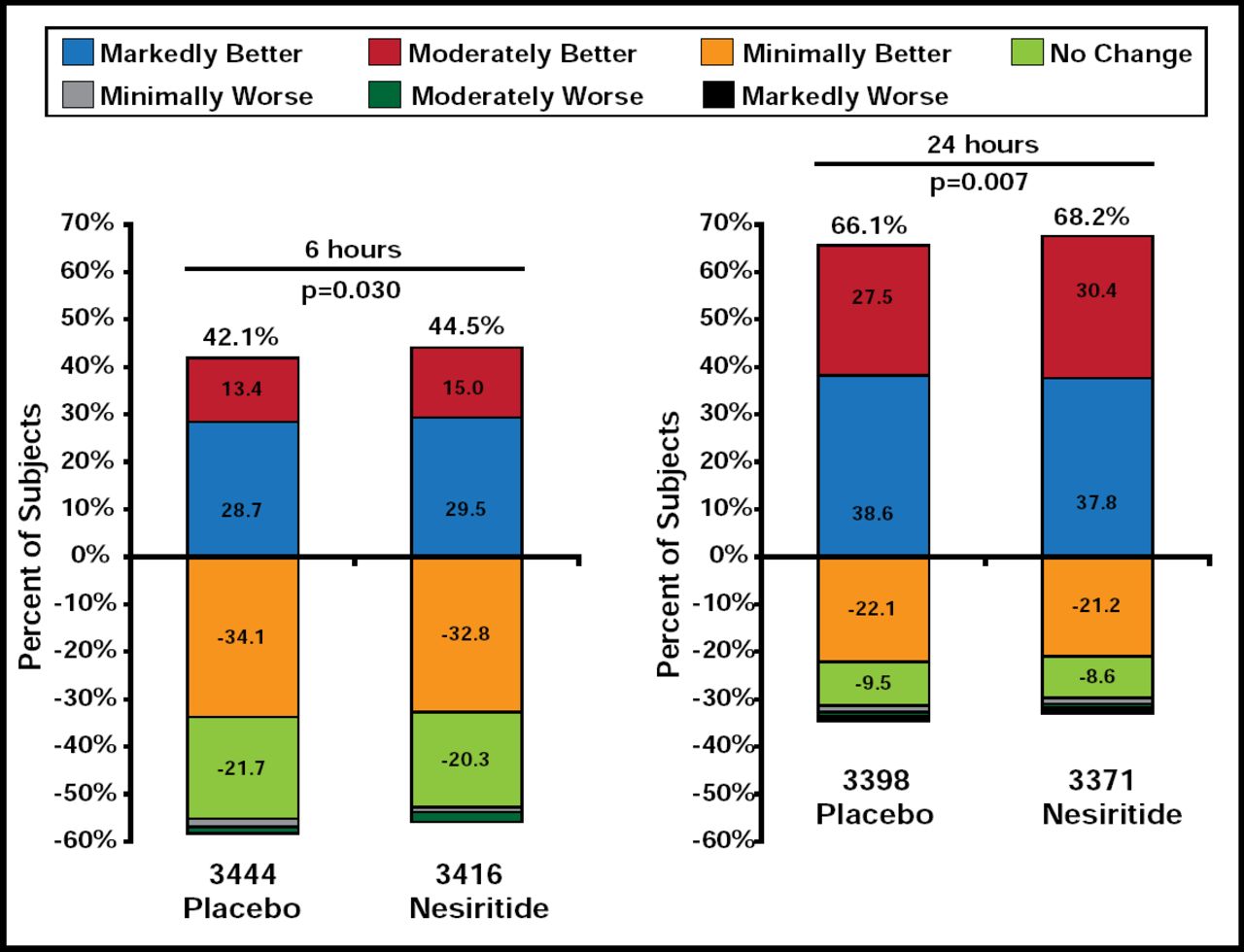
The two coprimary endpoints were rehospitalization for HF/all-cause mortality within 30 days and dyspnea at 6 or 24 hours (p-value for significance prespecified at ≤0.005 for both assessments or ≤0.0025 for either assessment). Improvement in dyspnea was self-reported using a 7-point Likert scale: markedly worse, moderately worse, minimally worse, no change, minimally better, moderately better, and markedly better. Safety endpoints included impact on renal function (25% decrease in eGFR through Day 30) and hypotension.
There was no difference in either of the coprimary endpoints between treatment groups. The rate of HF rehospitalization or 30-day all-cause mortality was 9.4% for subjects who were treated with nesiritide versus 10.1% for placebo-treated patients (HR, 0.93; 95% CI, 0.81 to 1.08; p=0.31). The observed reduction in overall dyspnea was modest and did not meet the preestablished criteria for significance. At 6 hours, 44.5% of nesiritide subjects reported markedly or moderately better dyspnea versus 42.1% of subjects who received placebo (p=0.030). At 24 hours, the rates were 68.2% for nesiritide patients versus 66.1% for placebo patients (p=0.007; Figure 1). Marked improvement in dyspnea was less frequent with nesiritide at 6 hours (15.0% vs 13.4%; p=0.03) and 24 hours (30.4% vs 27.5%; p=0.007). Neither finding was statistically significant, based on the predefined significant p-value of ≤0.005.

Coprimary Endpoint: 6- and 24-Hour Dyspnea.
Reproduced with permission from A. Hernandez, MD.
There was no difference in renal function between treatment groups (p=0.11). Subjects who received nesiritide experienced significantly (p<0.001) more hypotension compared with those who received placebo through Day 10 or discharge (HR, 11.3; 95% CI, 9.4 to 13.1).
- © 2010 MD Conference Express
Tools









{kind=link}
Table of contents
Cited By...
- No citing articles found.