

Summary
Sudden cardiac death (SCD) is a leading cause of death in the United States (amounting to 1 death every 80 seconds, or 450,000 deaths annually), with >50% of these events having a genetic etiology [Behr ER et al. Eur Heart J 2008]. There are several syndromes that are associated with SCD in the young, including congenital long QT syndrome, hypertrophic cardiomyopathy, arrhythmic right ventricular dysplasia, Brugada syndrome, and cathecholamine polymorphic ventricular tachycardia. This article discusses ways to identify these high-risk syndromes and current clinical approaches to SCD prevention in the young.
- Cardiology Clinical Trials
- Prevention & Screening Genomics
Sudden cardiac death (SCD) is a leading cause of death in the United States (amounting to 1 death every 80 seconds, or 450,000 deaths annually), with >50% of these events having a genetic etiology [Behr ER et al. Eur Heart J 2008]. There are several syndromes that are associated with SCD in the young, including congenital long QT syndrome (LQTS), hypertrophic cardiomyopathy (HCM), arrhythmic right ventricular dysplasia (ARVD), Brugada syndrome (BrS), and cathecholamine polymorphic ventricular tachycardia (CPVT). Kent Stephenson, MD, Huntington Hospital, Huntington, New York, USA, discussed ways to identify these high-risk syndromes and current clinical approaches to SCD prevention in the young.
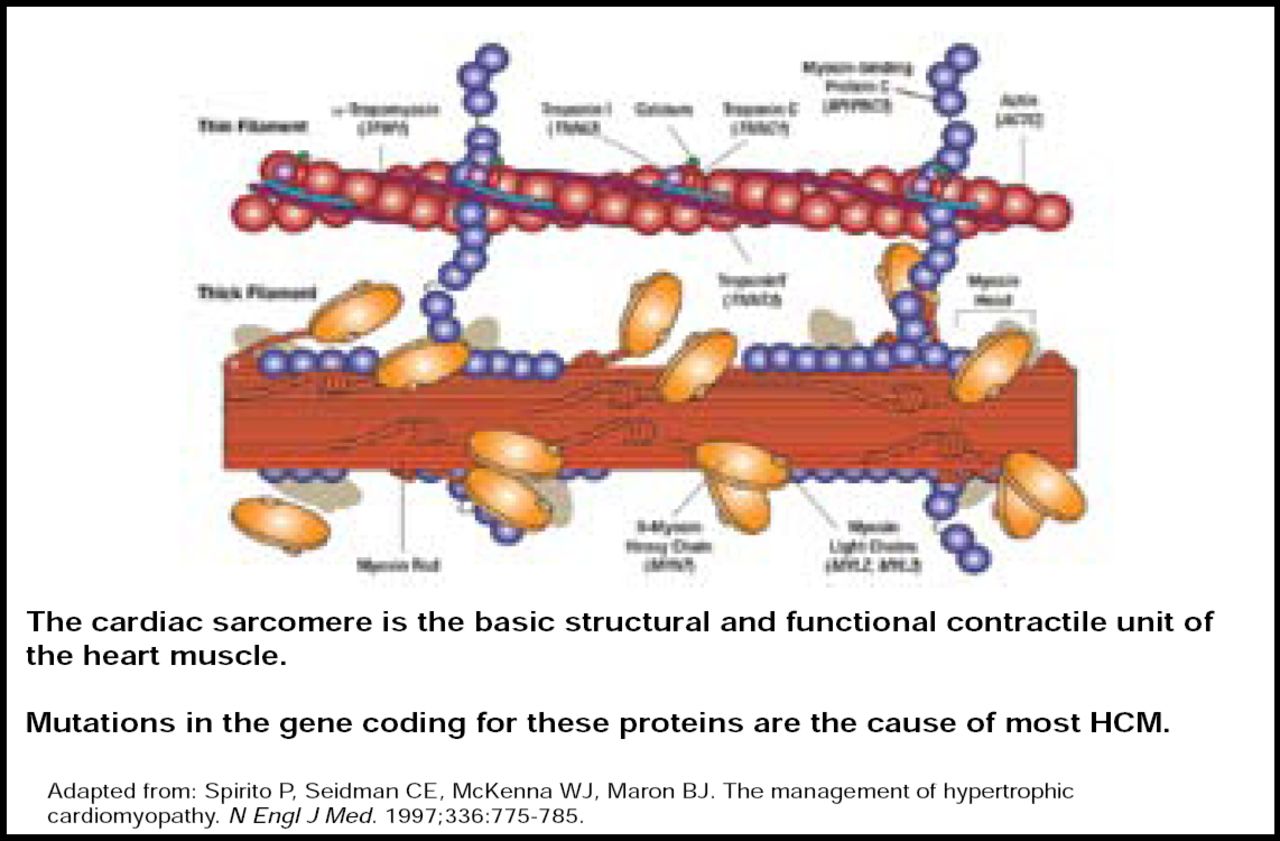
HCM is the major cause of SCD among athletes and people aged ≤30 years in the United States. It is the most common inherited cardiac disorder, with mutations occurring primarily within the cardiac sarcomere, particularly involving the β-myosin heavy chain (Figure 1) [Keren A et al. Nature 2008; Spirito P et al. N Engl J Med 1997]. HCM is characterized by unexplained thickening of the heart, and presentation varies from asymptomatic to severe limitation. Overall, mutation carriers tend to be younger, have greater left ventricular hypertrophy, and have a reverse curvature septal morphology (p<0.001) [Binder J et al. Mayo Clinic Proc 2006]. Calcium channel blockers, β-blockers, pacemakers, and implantable defibrillators are viable treatment options for HCM. However, in patients with obstructive symptoms, septal myomectomy is preferred over alcohol septal ablation (ASA), as ASA is associated with less favorable long-term outcomes [ten Cate FJ et al. Circ Heart Fail 2010].

Cardiac Sarcomere.
Copyright © 1997 Massachusetts Medical Society. All rights reserved.
BrS is a syndrome that is associated with SCD that often stems from genetic mutations of the SCN5A gene (which regulates the sodium channel), among others. The BrS phenotype is most commonly found in males aged 30 to 40 years with malignant arrhythmias and a history of syncope. BrS can be identified by the presence of coved- or saddleback-shaped ST-segment elevation in ECG leads V1 through V3, complete or incomplete right bundle branch block (RBBB), or T-wave inversion, but there are BrS mimics that may contribute to ST-segment elevation in the right precordial leads that should be considered [Wilde et al. Circulation 2002]. Potential treatments for BrS include quinidine to prevent recurrent syncope, isuprel to treat ventricular electrical storm, and implantable defibrillators for treatment of ventricular tachycardia/fibrillation and prophylaxis of sudden cardiac death. LQTS is characterized by delayed repolarization of individual action potentials and ECG, potentially resulting in QT interval prolongation and subsequent tachyarrhythmias [Roden DM et al. J Cardiovasc Electrophysiol 1999]. The most common LQTS genotype that is associated with sudden cardiac arrest at age <40 years is LQT1, though LQTS patients maintain a high risk for life-threatening cardiac events even after age 40. β-blockers are effective for treatment of LQTS, particularly among patients with LQT1 and LQT2 genotypes (p<0.001) [Moss AJ et al. Circulation 2000].
Catecholaminergic polymorphic ventricular tachycardia (CPVT) is a genetically transmitted cardiac ion channel disorder with an extremely high risk of SCD. If untreated, approximately 50% of individuals will present with a cardiac event that is possibly lethal by the age of 30 years. First recognized in 1975, this disorder involves mutations that lead to loss of calcium homeostasis in the heart, resulting in malignant arrhythmias.
The most common symptom is syncope, usually during periods of emotional or exertional stress. Syncope most often appears during the first or second decade of life. It is important to distinguish this from LQTS, since β-blocker therapy in individuals with this syndrome is less effective. In addition, the cardinal finding of prolongation of the QT interval that is seen in LQTS is absent in this disorder [Liu N et al. Progress in Cardiovascular Diseases 2008].
ARVD is a genetically determined disease of the heart muscle that is associated with arrhythmia, heart failure, and SCD. It is thought to be most common cause of SCD in the young in European countries. Unfortunately, SCD is often the first clinical manifestation of ARVD, especially among young people who are engaged in strenuous activity. While ARVD is often characterized by intramyocardial RV fatty infiltration, as seen on MRI, Dr. Stephenson cautions that reliance on this diagnostic indicator alone is not sufficient. Therefore, it is important to determine the diagnosis of ARVD, based on the composite of a number of possible findings, including ECG abnormalities (T-wave inversion in V1 to V3, RBBB, and/or epsilon wave), right ventricular structural abnormalities that are seen on imaging, abnormal myocardial biopsy, positive family history, and genetic testing. No one criterion is robust enough to adequately diagnose ARVD with confidence.
Detecting an increased risk of SCD in young patients is a clinical challenge, and often this risk is not detected until an event occurs, at which point it may be too late. However, there are various syndromes that may serve as early indicators of SCD risk. Thus, identifying these predictive markers is crucial to early detection and risk reduction. Recognizing the ECG footprints of these disorders is paramount to making a diagnosis in the asymptomatic individual. Genetic testing may also provide valuable prognostic and diagnostic data, as genetic mutations play a large role in SCD in the young.
- © 2010 MD Conference Express
Tools









{kind=link}
Table of contents
Cited By...
- No citing articles found.