

Summary
Numerous recent studies of animal models suggest a highly active role for the central nervous system in energy homeostasis. While this is not surprising, an expansion of these findings indicates that insulin signaling has a pronounced influence on the lifespan of a wide array of organisms.
- insulin
- endocrinology
Numerous recent studies of animal models suggest a highly active role for the central nervous system (CNS) in energy homeostasis. While this is not surprising, an expansion of these findings indicates that insulin signaling has a pronounced influence on the lifespan of a wide array of organisms. Jens Claus Brüning, MD, University of Cologne, Cologne, Germany, and winner of the 2008 Oskar Minkowski Prize, discussed these data in the 43rd Minkowski Lecture.
To the frustration of some researchers, insulin has been shown to have pleiotropic effects in nearly all tissues. Yet to date, the primary focus of these investigations has been on insulin activity in the peripheral tissues. Indeed, for many years insulin also was the primary hormone, or signaling molecule under consideration, but there was a paradigm shift in the understanding of metabolic regulation with the discovery and eventual isolation of leptin.
Leptin created a sensation in the lay press as well as in the peer-reviewed literature, because its activity in the body affected feeding behavior. Mice with defective leptin expression were observed to be obese (Shang et al. Nature 1994). Leptin deficiency in humans also was identified and fueled the assumption that the cause of obesity had indeed been found. Although patients did respond dramatically to leptin augmentation, the underlying hormonal defect was determined to be fairly rare. Research then shifted in perspective to consider not only the signal, but also the receiver. Leptin that is produced in adipose tissue has been shown to bind to the ventromedial nucleus of the hypothalamus, the center of appetite control. The message that is carried by this diminutive 16-kD protein is that the body has met its energy requirements, stimulating the brain to send signals to the body to stop eating.
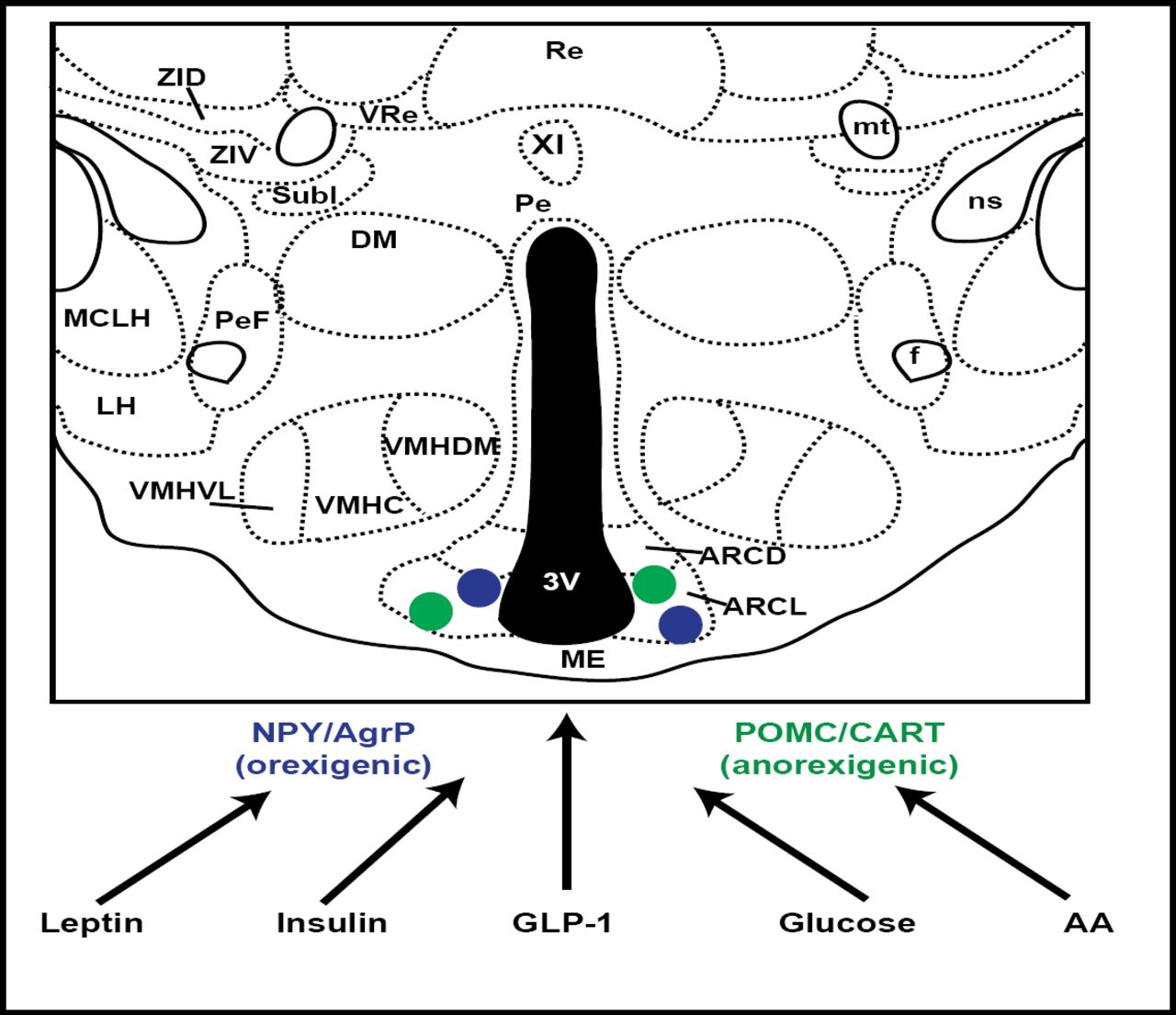
This activity was further localized to the ARC (hypothalamic arcuate nucleus) neurons, and thereafter multiple signals were identified as being targeted to this area. These included leptin, insulin, GLP-1, and glucose (Figure 1). The focus on ARC neurons was further validated by murine investigations, showing that if agouti-related peptide and neuropeptide Y, normally expressed in ARC, were disabled, feeding behavior was unaltered. However, if the neurons that expressed these genes were themselves ablated, food intake was drastically reduced, nearly to the point of starvation. ARC neurons are now known to play a critical role in the regulation of energy homeostasis (Gropp et al. Nat Neuro 2005).

ARC Neurons as Targets of Multiple Signals.
Insulin signaling in peripheral tissues entails activation of the insulin receptor substrate (IRS)-phosphatidylinositol 3-kinase (PI3K) enzyme system. In the hypothalamus, insulin functions with leptin as an afferent adiposity signal that is important for the regulation of body fat stores and hepatic glucose metabolism. Studies have shown that the IRS-PI3K pathway is a mediator of insulin action in the arcuate nucleus as well. Combined with recent evidence that leptin activates PI3K signaling in the hypothalamus, this suggests a conduit for neuronal crosstalk between insulin and leptin signaling (Niswender et al. Diabetes 2003).
Mice that lack IR have proven to be invaluable in gaining a better understanding of the role of insulin action in the brain. Downregulation of neuronal IR expression results in severe hyperinsulinemia and a dramatic upregulation of hepatic leptin receptor expression. Leptin replacement restored normal glucose metabolism in these mice. Insulin action, specifically in AgRP-expressing neurons, does play a critical role in controlling hepatic glucose production and may provide a target for the treatment of insulin resistance in type 2 diabetes (Koch et al. JCI 2008; Konner et al. Cell Metab 2007).
Caloric restriction has been strongly associated with a reduction in aging-associated pathologies, thereby resulting in a longer, healthier life. As first demonstrated in Drosophila (Chapman et al. Proc Roy Soc 1996), and later in C. elegans and mice, it is clear that restricted caloric intake can increase longevity.
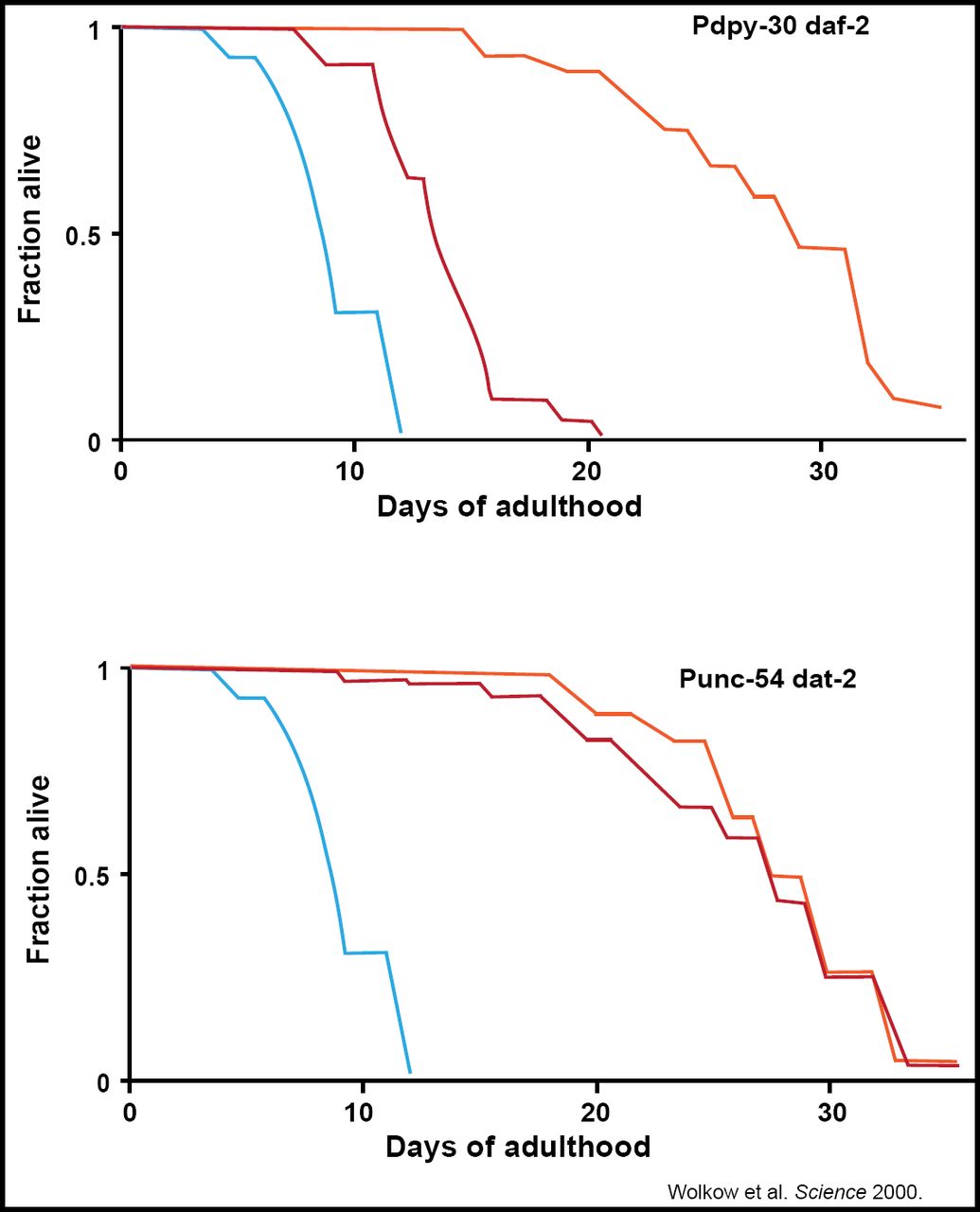
Selman et al. were able to localize this benefit to the CNS, observing that IRS-1-deficient mice stay healthy and disease-free longer and had the ability to maintain glucose homeostasis (FASEB 2008). This work was replicated in C. elegans, verifying that the nervous system's relationship with insulin is a central regulator of animal longevity (Wolkow et al. Science 2000; Figure 2).
The residence of such activity in the CNS raises a question about whether glucose dysregulation plays a role in age-related diseases of the brain, such as Alzheimer disease (AD). An animal model of AD was shown to have downregulated IGF-1R and IR and an excessive presence of their key substrate adaptor proteins, IRS-1 and IRS-2, the physiological equivalent of insulin resistance. The increase in IGF-1R was detected around and within amyloid beta-containing plaques, the pathological hallmark of AD, and resistant to IGF-1R/IR signaling (Moloney et al. Neurobiol Aging 2008); yet, this link is merely hypothesis-generating. One recent study by Moroz et al. assessed AD-type neurodegeneration in a type 2 diabetes mellitus (T2DM) mouse model but did not observe an AD histopathology, leading the authors to conclude that obesity and T2DM may contribute, but are not sufficient, to cause AD (Moroz et al. J Alzheimer's Disease 2008).

Restoring IIS in the CNS Abrogates the Life-Extending Effect of Systemic IR in C elegans.
The physiological importance of this homeostatic control system is highlighted by the severe obesity that results from the dysfunction of any of its key components. This new information provides a biological context within which to consider the global obesity epidemic and identifies numerous potential avenues for therapeutic intervention and future research (Schwartz et al. Nature 2006).
- © 2008 MD Conference Express
Tools









{kind=link}
{kind=link}
Table of contents
Cited By...
- No citing articles found.