

Summary
Research by the French physiologist Claude Bernard more than a century ago established the connection between the brain and the control of glucose levels, the main mechanism of glucose homeostasis and energy balance. Beginning with Professor Bernard's groundbreaking findings, This article discusses the history of hypothalamic research including his own current work on the impact of the hypothalamus on diabetes.
- Hyperglycemia/Hypoglycemia
- Endocrinology
- Diabetes & Metabolic Syndrome
- Hyperglycemia/Hypoglycemia
Research by the French physiologist Claude Bernard more than a century ago established the connection between the brain and the control of glucose levels, the main mechanism of glucose homeostasis and energy balance. Beginning with Professor Bernard's groundbreaking findings, Joel K. Elmquist, DVM, PhD, University of Texas Southwestern Medical Center, Dallas, Texas, USA, discussed the history of hypothalamic research including his own current work on the impact of the hypothalamus on diabetes.
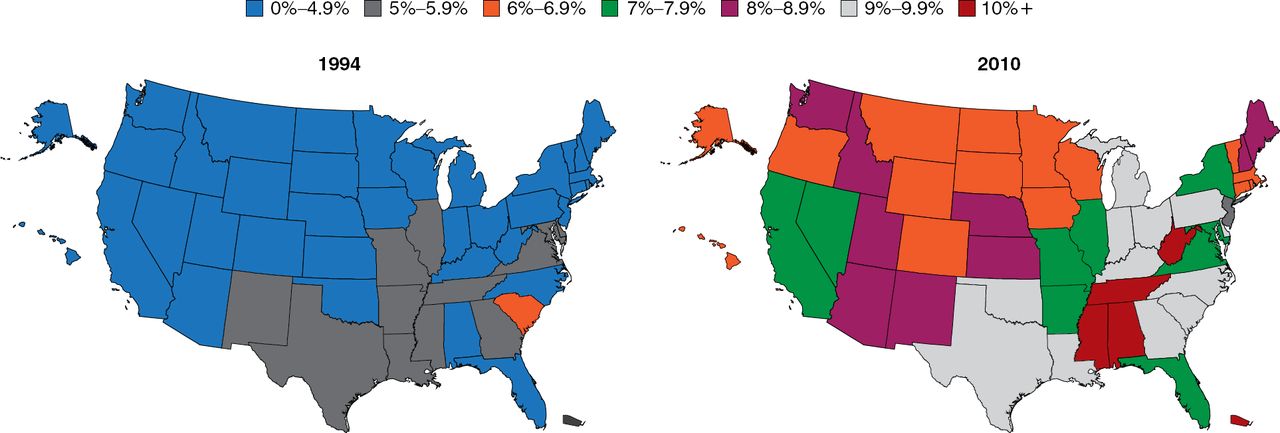
The prevalence of diabetes is increasing at an alarming rate (Figure 1). Particularly troubling is the increasing rate among children, which is occurring hand-in-hand with the rising rate of obesity. Among youths aged 2 to 19 years, 31.8% are overweight and 16.9% are obese [Dabelea D et al. JAMA 2014]. There is not an organ system in the body that is not adversely affected by the pathophysiology of diabetes; thus, this increase is truly a public health crisis, said Dr. Elmquist.

Increasing Prevalence of Type 2 Diabetes
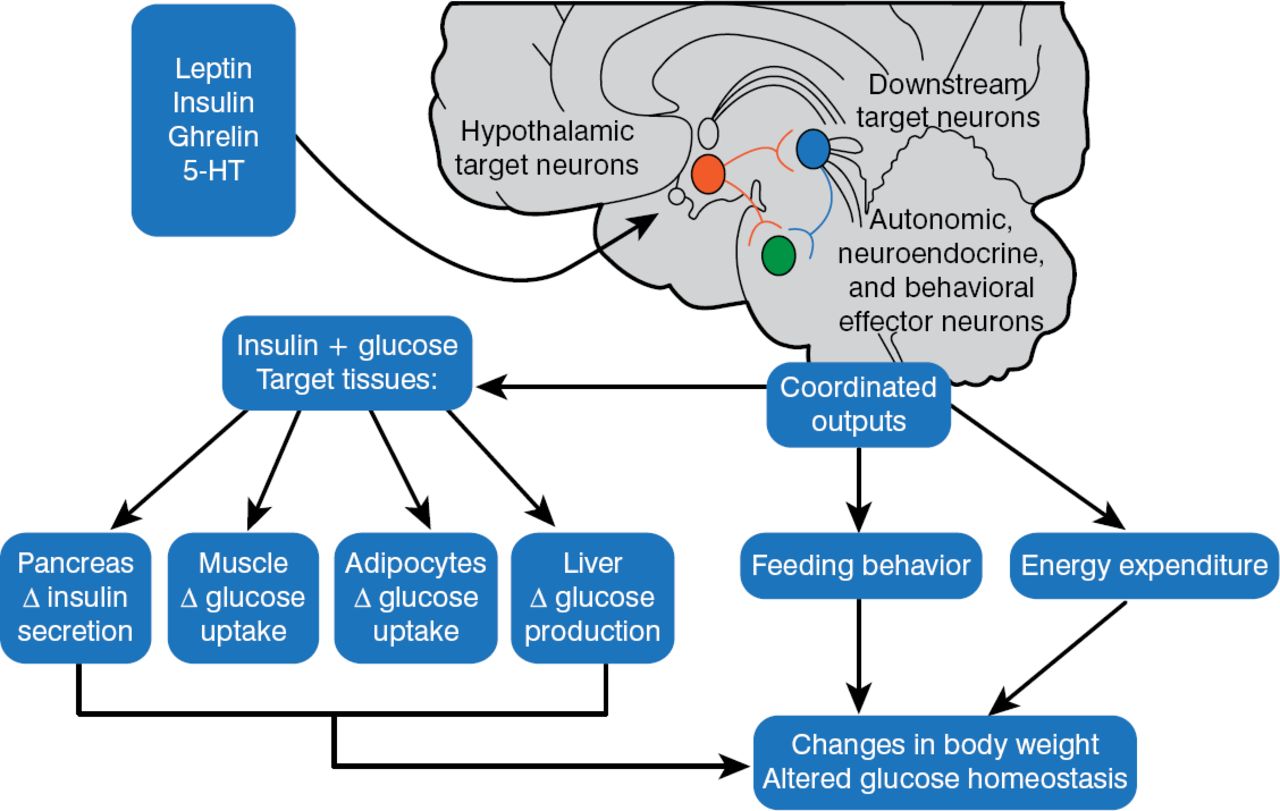
In 1940, Hetherington and Ranson published a key paper showing that the hypothalamus was critical for regulating food intake, body weight, and glucose homeostasis in rats. These same pathways were also found to be operational in humans. The 1997 discovery that melanocortinergic neurons, particularly melanocortin-4 receptors (MC4R) [Farooqi IS et al. J Clin Invest 2002], were associated with feeding and the agouti obesity syndrome in mice led to the identification of a genetics connection between obesity, the regulation of energy balance, and glucose homeostasis. Injection of recombinant human leptin in obese children with leptin deficiency produced sustained, beneficial effects on appetite, fat mass, hyperinsulinemia, and hyperlipidemia, confirming a relationship between phenotypic abnormalities and obesity in humans [Farooqi IS et al. J Clin Invest 2002]. Further, children with MC4R deficiency have a distinct obesity syndrome that is defined by a correlation between the signaling properties of these mutant receptors and energy intake [Farooqi IS et al. N Engl J Med 2003]. These observations affirmed research findings in mice and helped map out key pathways for diabetic pathology (Figure 2).

Pathways for Diabetic Pathophysiology
HT = hydroxytryptamine.
Adapted from Elmquist JK, Marcus JN. Nat Med 2003.
The current model of energy balance is viewed as a neuronal system in the brain that regulates energy intake, energy expenditure, and endogenous glucose production. The system senses and responds to input from hormonal and nutrient-related signals that promote energy homeostasis and the maintenance of blood glucose levels in the normal range. Defects in the system lead to obesity and type 2 diabetes mellitus (T2DM) [Schwartz MW, Porte D Jr. Science 2005].
Much attention has been focused on the melanocortin pathway in the brain and the peptide proopiomelanocortin (POMC) located in neurons of the hypothalamus, which act as endogenous ligands for MC4R. Pharmacologic manipulation of POMC gene expression could provide a potential way to combat obesity. For instance, α-MSH (melanocyte stimulating hormone), cleaved from a POMC precursor, decreases food intake, lowers body weight, and decreases blood sugar when given to animals. POMC neurons also activate leptin receptors and inhibit agouti-related peptides (AgRP), which positively influence feeding behavior.
The complexity of the POMC system led Dr. Elmquist and colleagues to try neuron-specific gene manipulation that allowed direct testing in awake, unrestrained mice, eg, deletion of genes expressing the Cre and flox genes. It is also possible to re-express genes in selected neurons, eg, the nucleus-specific re-expression of the ObR (leptin receptors that exhibit an obese phenotype). Mice with no ObR genes are prone to obesity, diabetes, infertility, and hypoactivity.
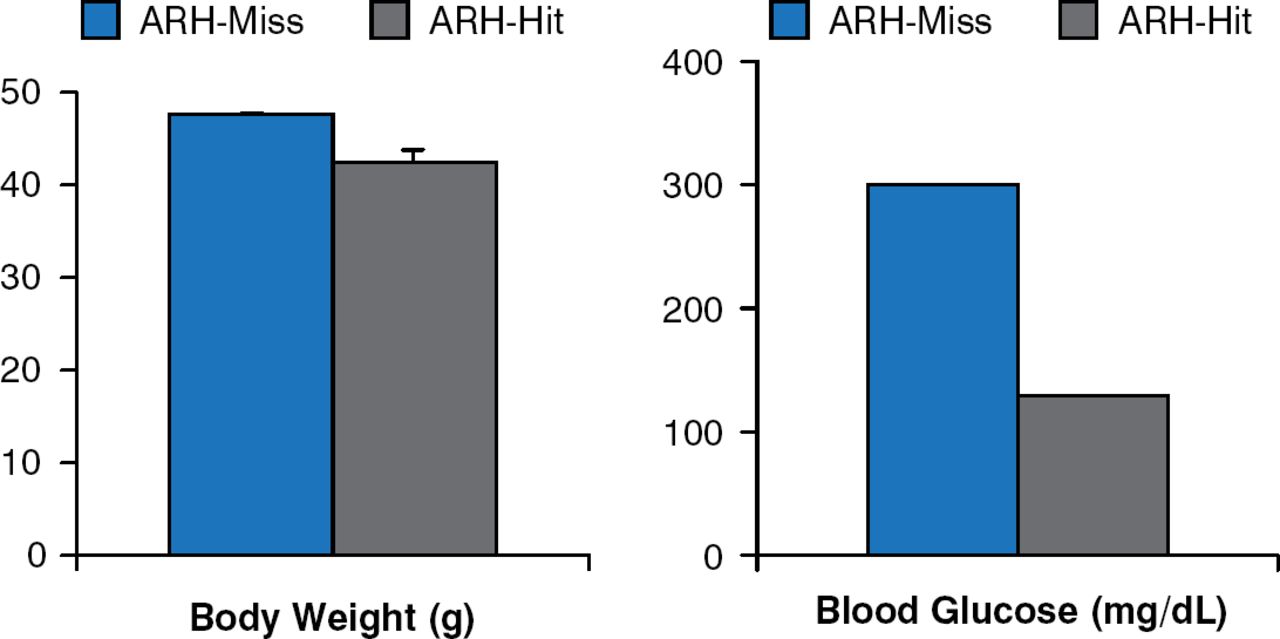
Leptin receptor re-expression with viral vectors in the hypothalamic arcuate nucleus (ARH) produces modest decreases in body weight and food intake. However, blood glucose levels are markedly improved, if not normalized (Figure 3) [Coppari R et al. Cell Metab 2005].

Leptin Receptor Re-expression in Hypothalamic Arcuate Nucleus Neurons
ARH = hypothalamic arcuate nucleus.
Reproduced from Coppari R et al. The hypothalamic arcuate nucleus: A key site for mediating leptin's effects on glucose homeostasis and locomotor activity. Cell Metab 2005;1:63–72. With permission from Elsevier.
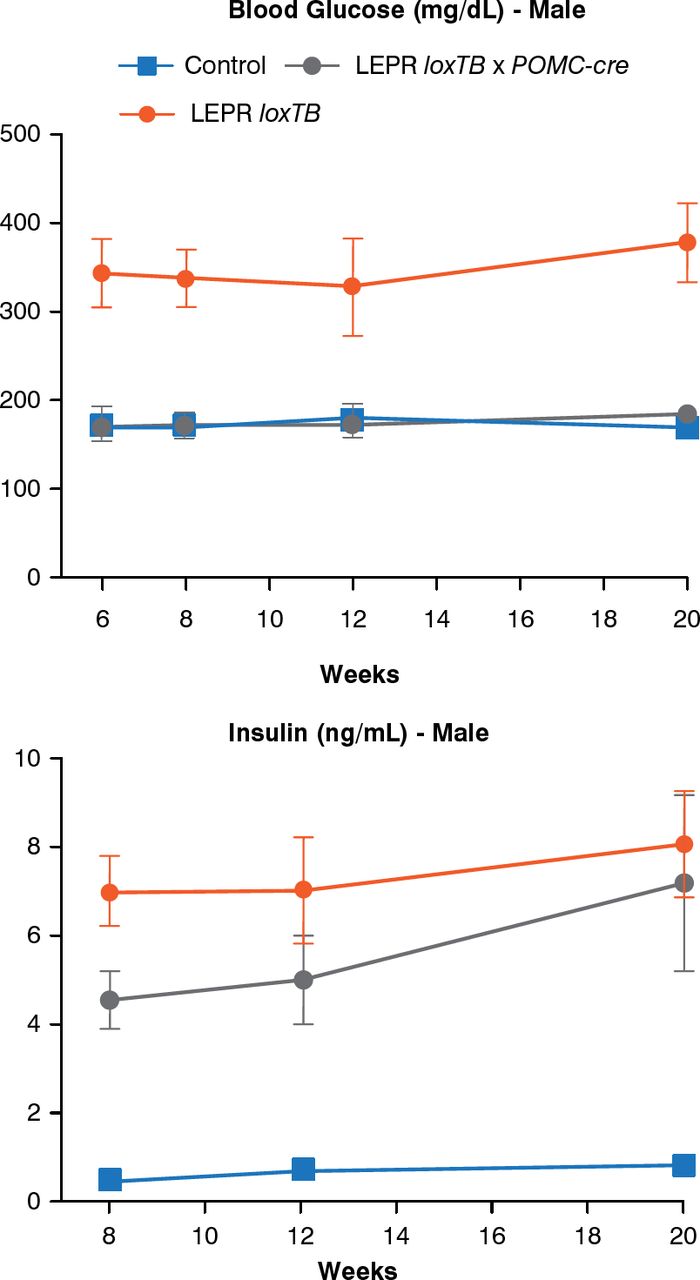
Another model can assess the role of leptin receptors in chemically identified neurons. In this model, endogenous leptin action on its receptor, prevented by a LoxP-flanked transcription blocker (loxTB), is then reactivated by Cre recombinase. Re-expression of leptin receptors only in POMC neurons in the ARH did not reduce food intake but partially normalized energy expenditure and modestly reduced body weight. Thus, it was concluded that leptin receptors in POMC neurons are not a major regulator of body weight or feeding [Berglund ED et al. J Clin Invest 2012]. However, leptin receptor re-expression in POMC neurons normalized blood glucose and ameliorated hepatic insulin resistance, hyperglucagonemia, and dyslipidemia. Thus, in the absence of obesity control, leptin can regulate glucose homeostasis (Figure 4).

Leptin Receptor Re-expression in POMC Neurons
LEPR = leptin receptor; loxTB = LoxP-flanked transcription blocker; POMC-cre = cre recombinase in POMC neurons.
Reproduced from Berglund ED et al. Direct leptin action on POMC neurons regulates glucose homeostasis and hepatic insulin sensitivity in mice. J Clin Invest 2012;122(3):1000–1009. With permission from the American Society for Clinical Investigation.
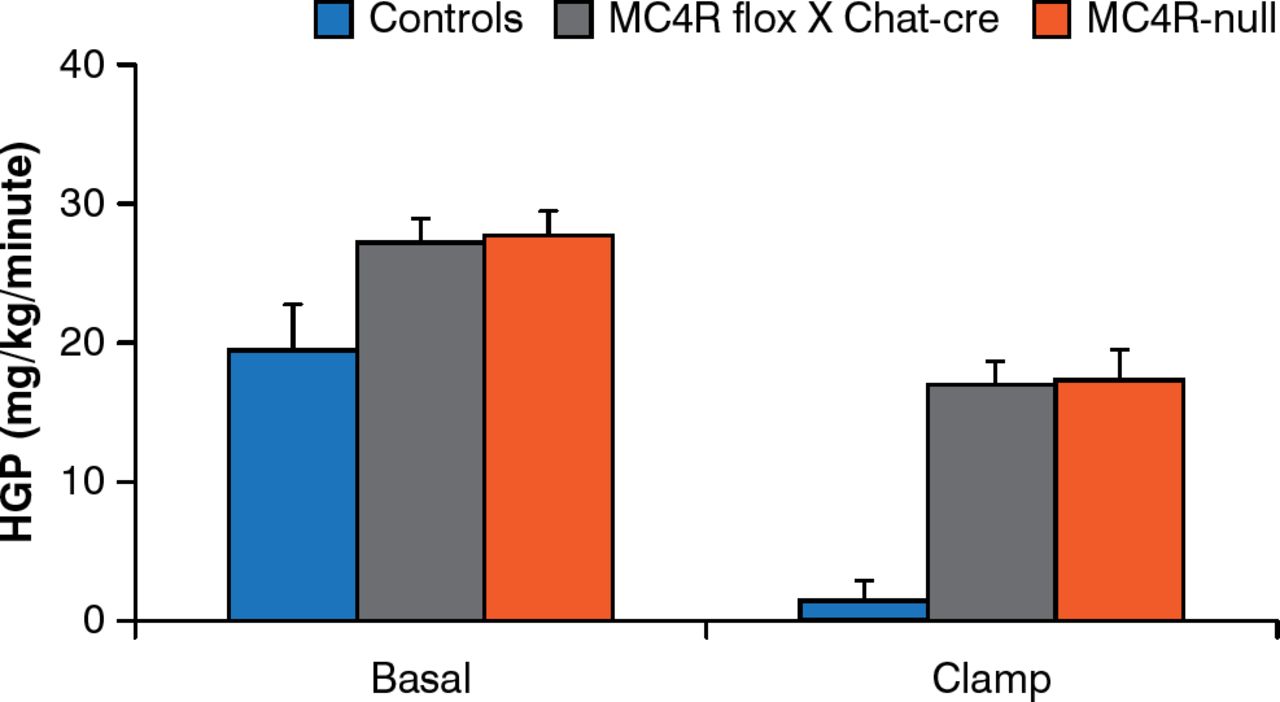
Brainstem neurons that control food intake and autonomic neurons that control energy expenditure are not the same. MC4Rs re-expressed by cholinergic (autonomic) neurons normalize hepatic glucose production but this is not true for MC4Rs expressed by brainstem neurons [Rossi J et al. Cell Metab 2011]. In addition, deleting MC4Rs in both sympathetic and parasympathetic cholinergic neurons increases food intake, the incidence of obesity, and impairs hepatic glucose homeostasis in mice (Figure 5) [Berglund ED et al. Nat Neurosci 2014].

Melanocortin-4 Receptors in Autonomic Neurons Regulate Hepatic Glucose Production
MC4R = melanocortin 4 receptors; controls = intact MC4R signaling; MC4R flox × Chat-cre = selective deletion of MC4R in cholinergic pre-ganglionic neurons; MC4R-null = ectopic ablation of MC4R; HGP = hepatic glucose production.
Reproduced from Berglund ED et al. Melanocortin 4 receptors in autonomic neurons regulate thermogenesis and glycemia. Nat Neurosci 2014;17(7):911–914. With permission from the Nature Publishing Group.
Thus, there are neurons (paraventricular nucleus of the hypothalamus) in the forebrain that express MC4Rs downstream of POMC and AgRP neurons and that are key regulators of food intake; sympathetic neurons regulate energy expenditure, glucose homeostasis, and hepatic insulin sensitivity, whereas vagus neurons regulate insulin secretion.
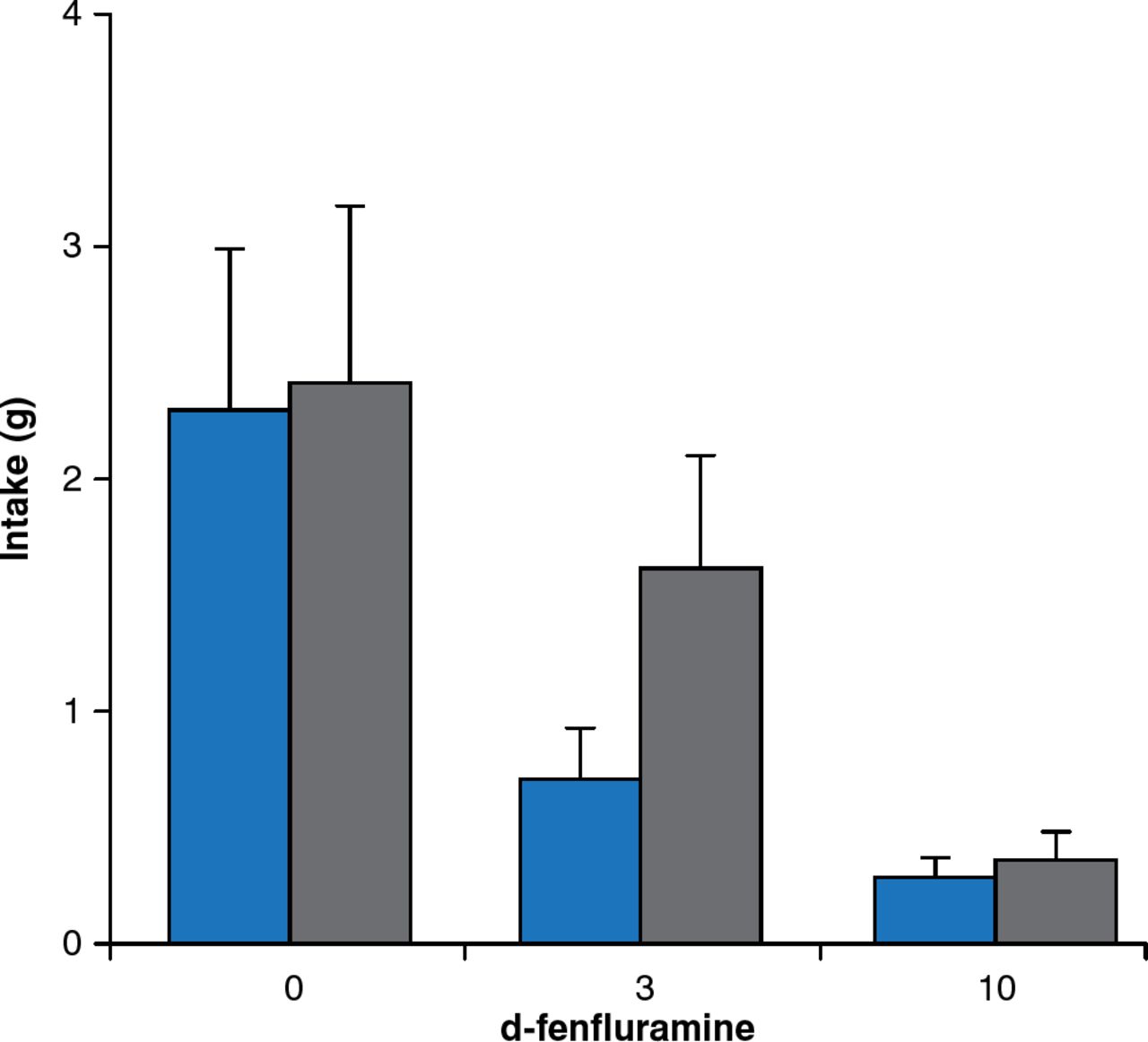
Besides leptin signaling, brain serotonin contributes substantially to the regulation of feeding and energy expenditure. Young adult mice with a targeted mutation of the serotonin 5-HT2C receptor gene consume more food despite normal responses to exogenous leptin administration. Mutant mice also respond more to high-fat feeding, leading to hyperglycemia without hyperlipidemia. The obesity drug fenfluramine increases serotonin release and inhibits reuptake, leading to significant weight loss. Mice lacking serotonin receptors are less sensitive to the satiating effects of d-fenfluramine 3 mg/kg compared with mice with intact serotonin receptors (Figure 6).

Blunted Satiety Response to d-Fenfluramine in Mice Lacking Serotonin Receptors
Gray bars = mice lacking serotonin receptors; blue bars = mice with serotonin receptors.
Reproduced from Vickers SP et al. Reduced satiating effect of d-fenfluramine in serotonin 5-HT2C receptor mutant mice. Psychopharmacology 1999;143:309–314. With permission from Springer Verlag.
Information generated from this research is translating into clinical studies. Leptin deficiency contributes to insulin resistance and is associated with severe lipodystrophy. Leptin-replacement has been shown to improve glycemic control and decrease triglyceride levels in patients with lipodystrophy and leptin deficiency [Oral EA et al. N Engl J Med 2002]. The selective serotonin 2C receptor agonist, lorcaserin, when given to obese individuals, is associated with a modest but significant weight loss compared with placebo [Smith SR et al. N Engl J Med 2010]. This class of compounds might be useful for the treatment of diabetes as well as obesity.
Claude Bernard was the first to think about how the brain controls complex metabolic pathways, particularly in the liver. Two hundred years later, he was more right than he knew. Going forward, a better understanding of these mechanisms will be very important for combating the growing epidemic of diabetes.
- © 2014 MD Conference Express®
Tools









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table of contents
Cited By...
- No citing articles found.