

Summary
The Paul Dudley White International Lecture was presented by Hugh C. Watkins, MD, PhD, Oxford, United Kingdom. Prof. Watkins described how the impact of genetics will grow and become a valuable tool for predicting efficacy, as well as aid in the development of treatments for individuals who are at risk of developing cardiovascular disease.
- Coronary Artery Disease
- Inflammatory Disease Genomics
The Paul Dudley White International Lecture was presented by Hugh C. Watkins, MD, PhD, Oxford, United Kingdom. Prof. Watkins described how the impact of genetics will grow and become a valuable tool for predicting efficacy, as well as aid in the development of treatments for individuals who are at risk of developing cardiovascular disease.
Both Mendelian mutations (that cause inherited diseases that run in families) and common susceptibility variants that have been identified through a genome wide association study (GWAS) improve our understanding of biology and have the potential to identify new drug targets. Mendelian mutations are rare but have a very large effect and are strongly predictive, while common variants have a small effect and are not very good for predicting individual risk of a given disease.
An understanding of Mendelian or inherited disease variants can lead to understanding causality and certain proof. As an example, genetic research showed that hypertrophic cardiomyopathy (HCM) was caused by mutations in sarcomeric protein genes—in particular, mutations in the thick and thin filament proteins that are associated with contraction. In vitro functional assays show that HCM mutations increase the Ca2+ sensitivity of contractility, whereas dilated cardiomyopathy mutations decrease it. Because troponin is the major Ca2+ buffer in the cardiomyocyte sarcoplasm, it has been suggested that Ca2+ affinity changes that are caused by cardiomyopathy mutant proteins may directly affect the transient Ca2+ and hence calcium handling and hypertrophy signaling [Robinson P et al. Circ Res 2007].
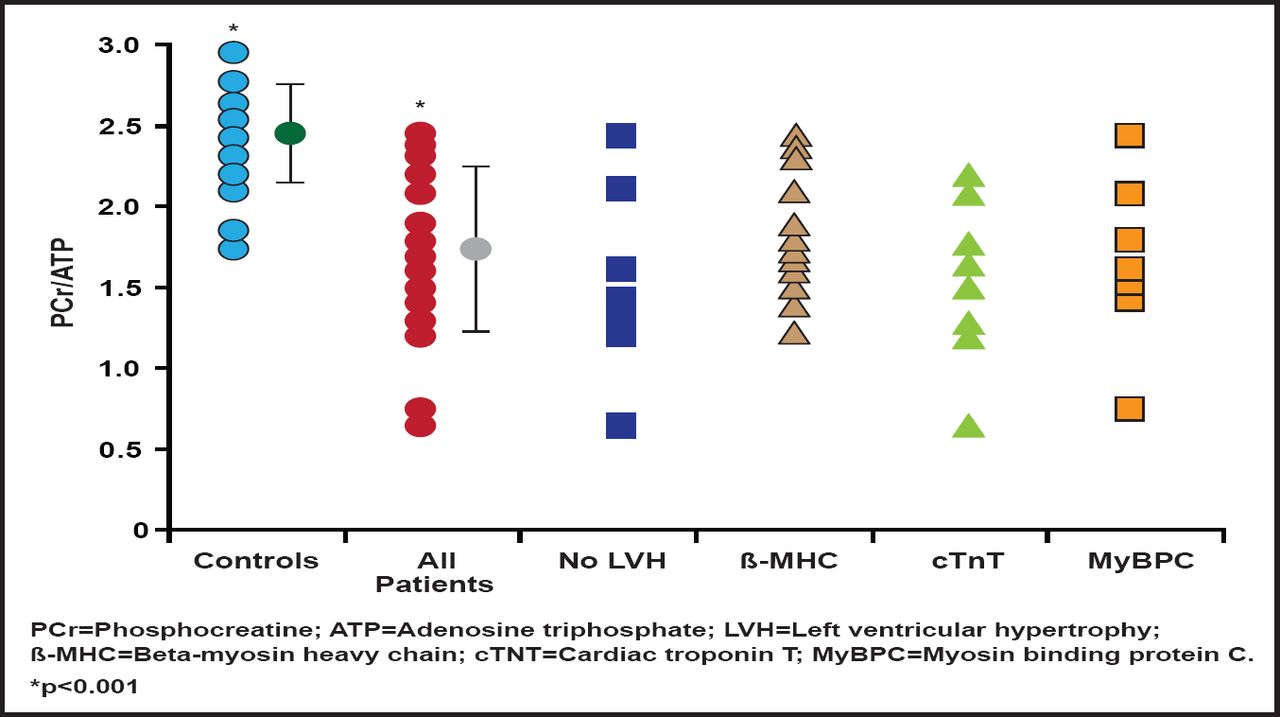
Further, the mutations that cause HCM increase activation of the heart muscle, increase the energy that is used to generate contraction, and deplete adenosine triphosphate (ATP). In a study that investigated cardiac energetics in subjects with mutations in three different familial HCM disease genes, some of whom were nonpenetrant carriers without hypertrophy, the cardiac phosphocreatine (PCr)-to-ATP ratio was reduced in the HCM subjects by ∼30% relative to controls in all three disease gene groups. The PCr/ATP ratio was equally reduced in subjects with and without left ventricular hypertrophy (Figure 1). This bioenergetic deficit in genotype-confirmed HCM, even in those without hypertrophy, supports a proposed link between altered cardiac energetics and development of the disease phenotype [Crilley J et al. J Am Coll Cardiol 2003]. Prof. Watkins pointed out new unpublished data that show that the preexisting energetic deficit in HCM is acutely exacerbated by exercise.

PCr/ATP Ratio.
Reprinted from Journal of the American College of Cardiology, 41 /10, Crilley J et al, Hypertrophic cardiomyopathy due to sarcomeric gene mutations is characterized by impaired energy metabolism irrespective of the degree of hypertrophy, Pages 1776–1782, Copyright 2003, with permission from the American College of Cardiology.
All of the sarcomeric mutations appear to result in inefficient use of ATP, suggesting that an inability to maintain normal ATP levels may be a central abnormality. In a study that examined candidate genes that are involved in energy homeostasis in the heart in families with a syndrome that comprises severe HCM with Wolff Parkinson White syndrome and atrioventricular conduction disease, mutations were discovered in PRKAG2, which encodes the gamma(2) subunit of AMP-activated protein kinase (AMPK). Since AMPK provides a central sensing mechanism that protects cells from exhaustion of ATP supplies, this finding substantiates energy compromise as a unifying pathogenic mechanism in HCM [Blair E et al. Hum Mol Genet 2001].
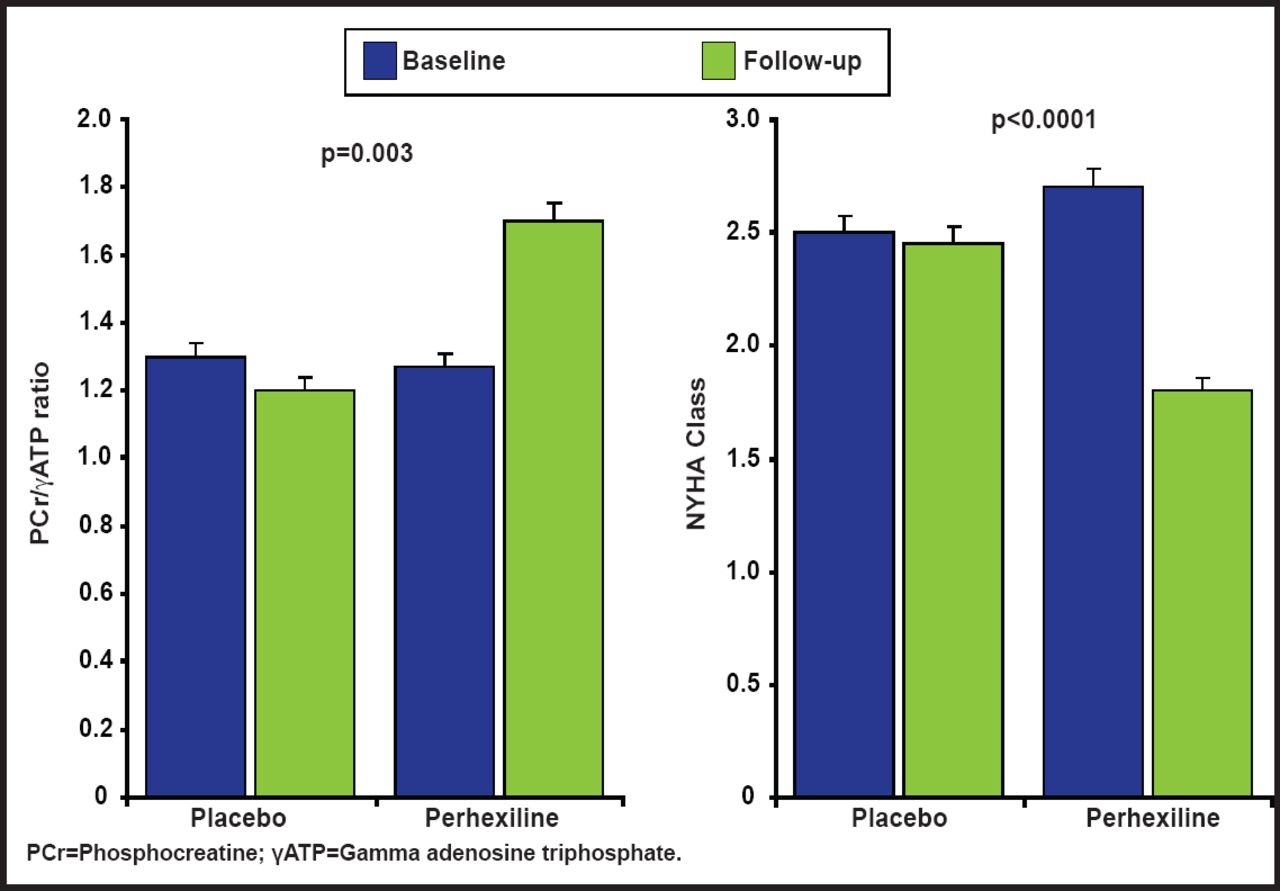
Clinical findings from a randomized, controlled trial support the pathophysiology that is described above. The investigators hypothesized that the metabolic modulator perhexiline would ameliorate myocardial energy deficiency and thereby improve diastolic function and exercise capacity in patients with symptomatic exercise limitations that are caused by nonobstructive hypertrophic cardiomyopathy. A study in 46 patients showed that perhexiline ameliorated cardiac energetic impairment, reduced heart failure symptoms (Figure 2), and increased exercise capacity [Abozguia K et al. Circulation 2010].

Randomized, Controlled Trial of Metabolic Modulation with Perhexiline in HCM.
Copyright © 2010 American Heart Association. All rights reserved.
Inherited cardiac conditions that are currently amenable to genetic testing include hypertrophic cardiomyopathy, arrhythmogenic right ventricular cardiomyopathy, and channelopathies (eg, long QT syndrome). Since mutations are not found in all affected patients, it is not possible to rule out the presence of the disease in patients without such mutations. In addition, genetic testing may not modify treatment in an individual who clearly meets phenotypic criteria for the condition. However, once a mutation is identified in a confirmed case, testing for that variant is useful in identifying other affected family members, particularly if the clinical diagnosis is difficult and expensive or if the presymptomatic diagnosis is clinically relevant. Utility and cost effectiveness have led to the inclusion of deoxyribonucleic acid (DNA) testing testing in the guidelines to determine who is at risk.
At the other end of the spectrum, the usefulness of common variants for predicting individual risk of common disease is not as clear. Some variants increase patient risk and some decrease risk. The effects are small, variants are numerous, and they explain a fraction of the estimated heritability. Most of these variants remain unknown, but in the case of coronary artery disease (CAD), more than 30 loci are now implicated.
To harness the novel findings from GWAS studies, there is a pressing need to know which gene confers susceptibility at each locus and whether its expression is up- or downregulated before testing interventions. When used as drug targets, the small size of the genetic effects matter little, as long as causality is clear and the pathway is logical. Uncovering these new loci will be beneficial and could lead, for example, to our understanding of new lipoprotein handling pathways.
A novel gene chip to test for genetic associations in subjects with coronary disease identified two lipoprotein(a) (LPA) variants variants that were strongly associated with both an increased level of Lp(a) and an increased risk of coronary disease. A meta-analysis showed that with a genotype score that involved both LPA SNPs, the odds ratios for coronary disease were 1.51 (95% CI, 1.38 to 1.66) for one variant and 2.57 (95% CI, 1.80 to 3.67) for two or more variants [Clarke R et al. N Engl J Med 2009]. Importantly, these findings strongly support a causal role for Lp(a) in CAD and indicate that lowering levels is a valid therapeutic target (as is achieved, for example, by CETP inhibitors).
In Prof. Watkins's opinion, “we need to be cautious regarding how much risk prediction benefit we can expect.” Our ability to predict common diseases, based on multiple genetic variants alone or in addition to traditional disease risk factors, has been limited thus far and is likely to remain so. Genetic profiling for personalizing medicine requires tests that fairly accurately predict disease risk, particularly when interventions are invasive or expensive or have major side effects [Janssens AC and van Duijin CM. Hum Mol Genet 2008], and these tests are not yet available. Instead, new GWAS findings are likely to have a major impact on our understanding of mechanisms of disease and, over time, new approaches to therapy.
- © 2011 MD Conference Express®
Tools









{kind=link}
{kind=link}
Table of contents
Cited By...
- No citing articles found.