

Summary
Arrhythmogenic right ventricular cardiomyopathy (ARVC), an inherited myocardial disorder, is associated with arrhythmias, heart failure, and sudden death [Syrris P et al. Am J Hum Genet 2006; Yang Z et al. Circulation Res 2006]. Clinical diagnosis of ARVC can be complicated, because multiple diagnostic tests are required and clinical presentation varies. This article discussed the etiology of ARVC and recent breakthroughs concerning detection, as well as the issue of hypertrophic cardiomyopathy.
- cardiology genomics
- inflammatory disease
Arrhythmogenic right ventricular cardiomyopathy (ARVC), an inherited myocardial disorder, is associated with arrhythmias, heart failure, and sudden death [Syrris P et al. Am J Hum Genet 2006; Yang Z et al. Circulation Res 2006]. Clinical diagnosis of ARVC can be complicated, because multiple diagnostic tests are required and clinical presentation varies. William McKenna, MD, University College London, London, UK, discussed the etiology of ARVC and recent breakthroughs concerning detection.
Desmosomes consist of several proteins that serve as specialized intercellular junctions of the cardiac and endothelial tissue that are involved in cell-to-cell adhesion and intracellular signaling. ARVC is a genetic disorder that involves desmosomal mutations, particularly in the desmoplakin, N-cadherin, and plakoglobin pathways. Within single families that carry these mutations, there are variations in ARVC presentation, such as age at onset, clinical features, and outcomes [Bauce B et al. Eur Heart J 2005; Norman M et al. Circulation 2005; Kannankeril PJ et al. Heart Rhythm 2006; Syrris P et al. Eur Heart J 2007].
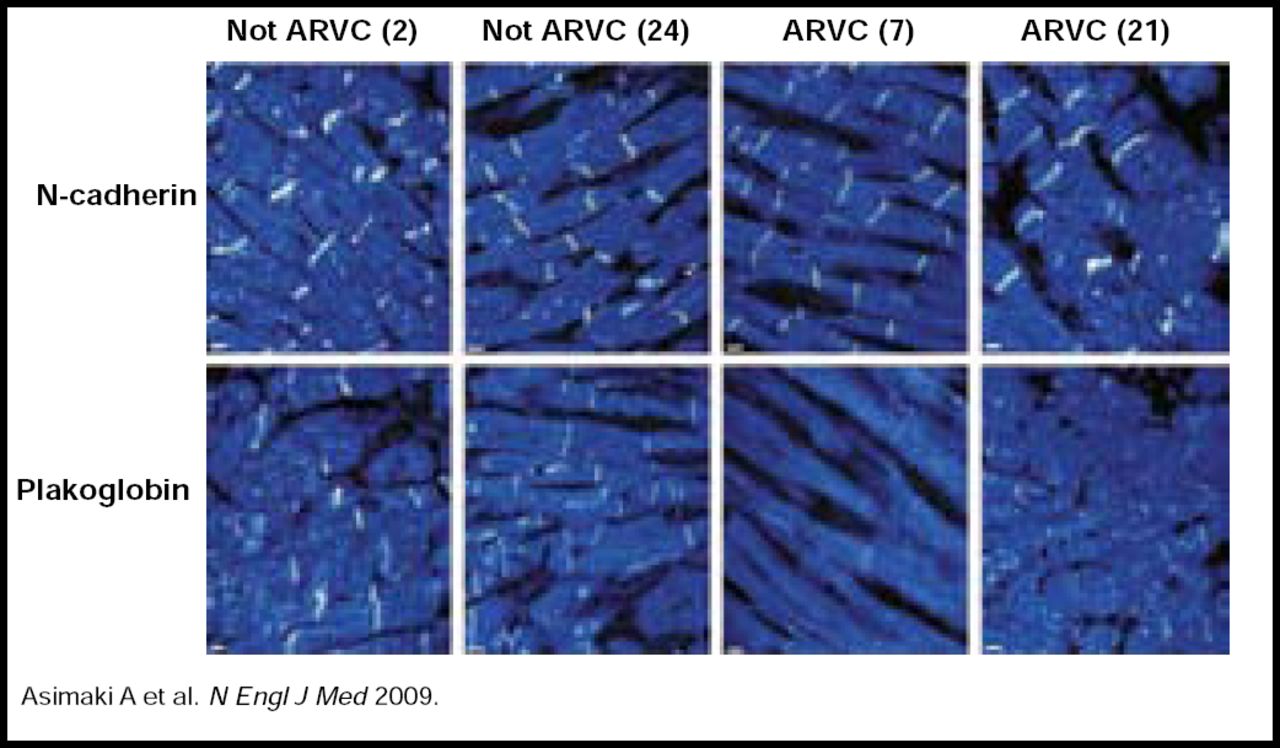
In a recent study by Asimaki and colleagues, routine immunohistochemical analysis of endomyocardial biopsy samples revealed high sensitivity (91%) and specificity (82%) for ARVC diagnostic testing (Table 1). Ten of 11 subjects were correctly diagnosed with ARVC based on clinical criteria, and AVRC was accurately ruled out in 10 of 11 subjects without ARVC (positive predictive value 83% and negative predictive value 90%; Figure 1) [Asimaki A et al. N Engl J Med 2009]. Prof. McKenna concluded that these recent findings and ongoing genetic studies may have practical implications for ARVC detection in the clinical setting.
Clinical and Immunohistological Diagnoses in Blinded Analysis of Cardiac Biopsy Specimens.

Immunofluorescence Images of Endomyocardial Biopsy Samples from Two Subjects with ARVC and Two Subjects without ARVC.
Copyright © 2009 Massachusetts Medical Society. All rights reserved.
Representative images from a blinded analysis of endomyocardial biopsy samples show that immunoreactive signal levels for n-cadherin and plakoglobin in two subjects with AVRC differ from the signal levels in two subjects without ARVC. Numbers in parentheses correspond to the subject numbers in Table 1.
Patrick O'Gara, MD, Brigham & Women's Hospital, Boston, MA, discussed the issue of hypertrophic cardiomyopathy (HCM), which also involves genetic mutations that lead to myocyte disarray and increased collagen production (sarcomere mutations, including β-MHC, MBP-C, and Tn; nonsarcomere mutations, including PRKAG2, LAMP-2, and glycogen storage disease). “There is extraordinary phenotypic variability associated with hypertrophic cardiomyopathy, making it more difficult to identify and treat,” said Dr. O'Gara.
Identification of HCM and potential obstruction consists of noninvasive imaging of myocardial structure as well as left ventricular outflow gradient assessment by either provoking a response by positioning (valsalva versus rest) in the clinical setting or performing exercise and imaging in the echocardiography lab. Outflow tract obstruction is present in 70% of patients with HCM, of which only 33% is revealed following exercise-related provocation. Identifying obstruction is clinically important, because its presence is associated with increased mortality, and such patients deserve closer clinical follow-up and consideration for more aggressive therapy [Maron M et al. N Engl J Med 2003]. Currently, there are no data available regarding pharmaceutical therapy and its impact on sudden cardiac death rates in these patients, Dr. O'Gara noted.
Possible interventional options for HCM patients include septal myectomy and alcohol septal ablation. Alcohol septal ablation involves creating an ethanol-induced myocardial infarction, while septal myectomy entails surgical removal of the hypertrophied basal septum. Myectomy has a 90% rate of success, while alcohol septal ablation has a slightly lower chance of successful outcome (70% to 80%). However, the safety of both of these interventions has been established (<2% to 3% mortality) [Nishimura R & Holmes D. N Engl J Med 2004; Maron BJ et al. Circulation 2007]. Septal myectomy is the preferred surgical intervention for HCM in the presence of concomitant problems, such as multivessel coronary disease, intrinsic mitral valve disease, midventricular obstruction, and fixed subaortic obstruction [Nishimura R & Holmes D. N Engl J Med 2004].
Dr. O'Gara concluded that HCM screening is important, especially for first-degree relatives. Screening should include examination, ECG, and echocardiography annually during adolescence (age 12 to 18 years) and every 3 to 5 years after age 18 years if echocardiograms are normal. When more than one family member is affected, further genetic testing may be indicated.
- © 2010 MD Conference Express
Tools









{kind=link}
Table of contents
Cited By...
- No citing articles found.