

Summary
The Robert Wartenberg Lecture is the premier award and address at the American Academy of Neurology's Annual Meeting. In 2014, the award was given to David M. Holtzman, MD, Washington University School of Medicine, St. Louis, Missouri, USA. Dr. Holtzman lectured on the current state of our understanding of biomarkers for Alzheimer's disease (AD) and potential immunotherapeutic approaches.
- Cognitive Disorders
- Dementias
- Prevention & Screening
- Cognitive Disorders
- Neurology
- Dementias
- Prevention & Screening
The Robert Wartenberg Lecture is the premier award and address at the American Academy of Neurology's annual meeting. This year, the award was given to David M. Holtzman, MD, Washington University School of Medicine, St. Louis, Missouri, USA. Dr. Holtzman lectured on the current state of our understanding of biomarkers for Alzheimer's disease (AD) and potential immunotherapeutic approaches.
There are 2 major types of AD: early-onset familial (<1% of cases) and late-onset (age >60 years; >90% of cases) AD. The strongest risk factors are age and genetics. AD progresses in stages from very mild to severe, on average, over an 8- to 12-year period. Clinical features include gradual onset and progression, memory deficits (particularly recent memory), cognitive dysfunction (eg, executive function, problem solving, attention, language skills), and behavioral dysfunction, such as personality change, depression, delusions, hallucinations, apathy, and sleep disruption. AD is now usually recognized in its early clinical phases on the basis of changes in previous levels of performance as recognized by an informant and validated by additional testing.
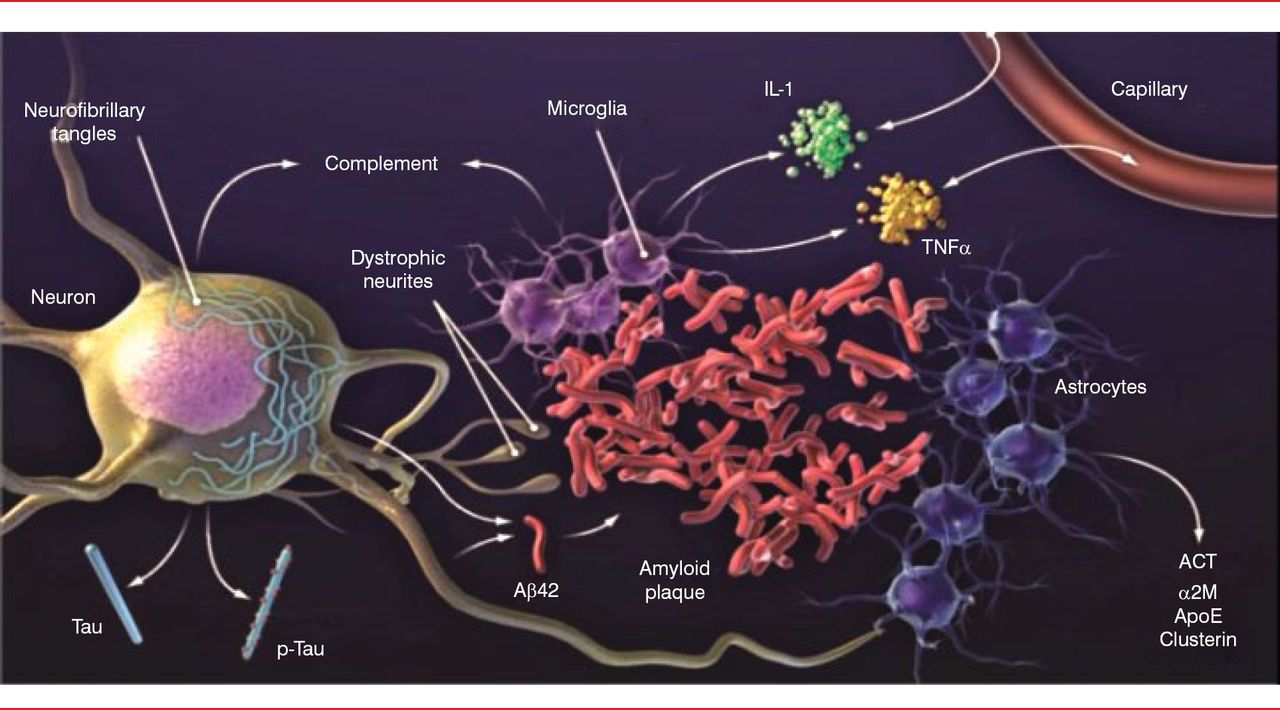
The classic neuropathophysiology of AD was first described in the early 1900s. The process begins with plaque buildup, followed by neurofibrillary tangles (tau) [Holtzman DM et al. Sci Transl Med 2011]. These changes are associated with nerve cell and synapse dysfunction, loss of connections, inflammation, brain shrinkage, and cell death. Our understanding of AD pathology has progressed a great deal in the past 25 years. The amyloid plaques are now known to be made up of the amyloid-β (Aβ) peptide surrounded by dystrophic neurites. The plaques are also surrounded by astrocytes and microglial cells that secrete a variety of cytokines and other molecules, including complement components, interleukin-1, tumor necrosis factor-α, α-1 antichymotrypsin, α-2 macroglobulin, apolipoprotein E, and clusterin. Neurons develop intracellular aggregates of tau in cell bodies and dendrites (Figure 1).

Brain Pathology of Alzheimer's Disease
Aβ=amyloid-β; ACT=antichymotrypsin; ApoE=apolipoprotein E; α2M=α-2 macroglobulin; IL-1=interleukin-1; TNFα=tumor necrosis factor-α.
Reproduced from Holtzman DM et al. Alzheimer's disease: the challenge of the second century. Sci Transl Med. 2011;3(77):77sr1. With permission from the American Association for the Advancement of Science.
Aβ and tau interact to contribute to the pathogenesis of AD. Aβ aggregation leads to local toxicity and inflammation and contributes to the exacerbations of tau accumulation. Once Aβ aggregates into oligomers and fibrils, cell toxicity, inflammation, and increased conversion of soluble tau into aggregated tau occur. The accumulation of tau spreads into other parts of the neocortex and contributes to the cognitive decline. There is evidence that Aβ aggregation is influenced by the Aβ-binding molecule apolipoprotein E, a strong genetic risk factor for AD.
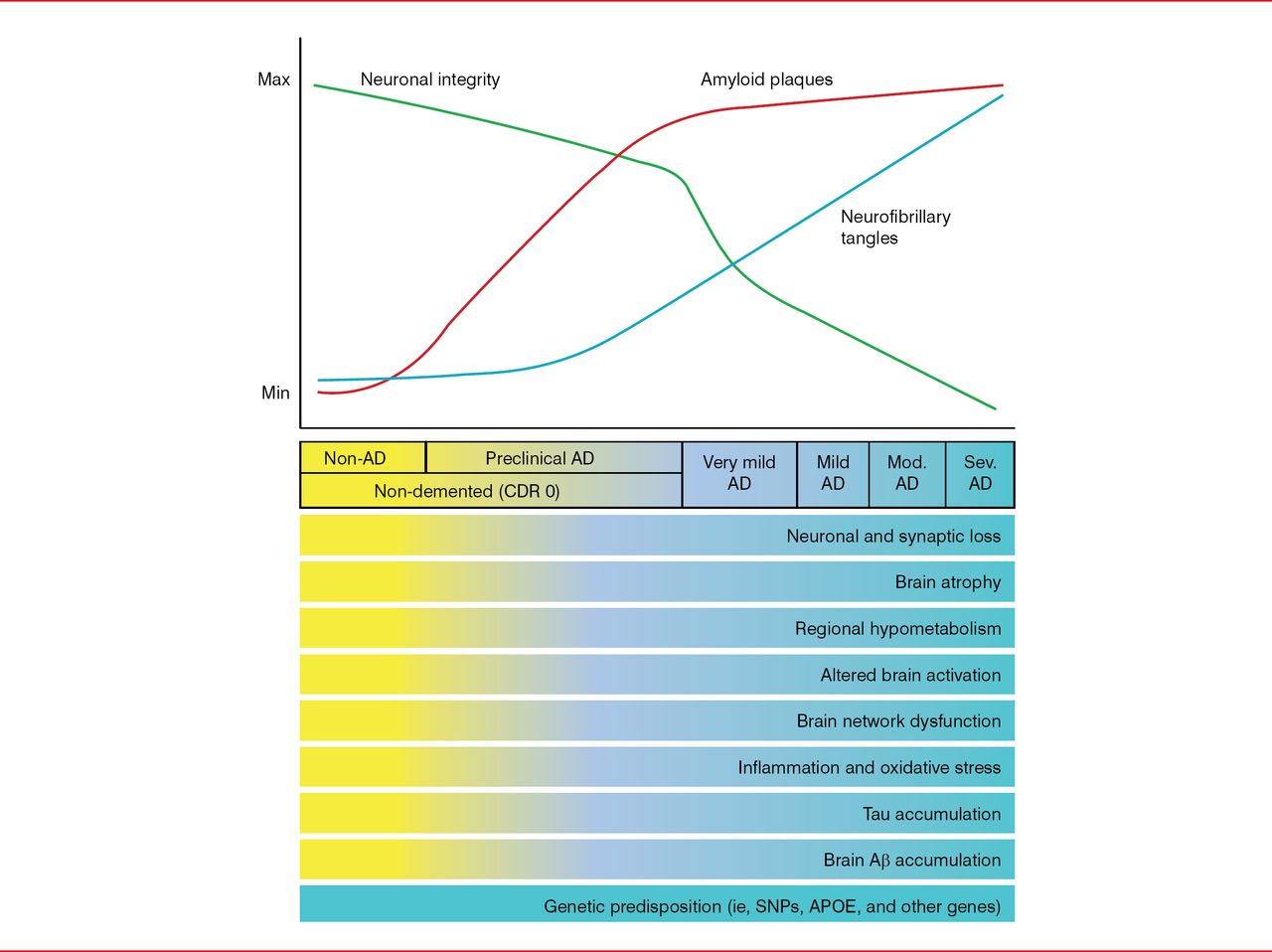
The time course of developing the classic AD-related pathologic changes in relation to developing clinical manifestations is important to understand. Pathologically, there is an initial quick increase of amyloid plaques that peaks early on and a gradual increase in neurofibrillary tangles that spreads into the temporal neocortex and peaks in later clinical stages of disease, accompanied by a linear decline in neuronal integrity. Aβ peptide aggregation begins about 15 years before the onset of memory loss and dementia. Tau elevation, reflecting neurodegeneration, probably begins about 5 years before the onset of symptoms (Figure 2). Thus, it appears critical to develop treatments that are effective before patients become symptomatic and significant damage to the brain has occurred.

AD Biomarker Changes Relative to Cognitive and Clinical Changes
Aβ=amyloid-β; AD=Alzheimer's disease; APOE=apolipoprotein E; CDR=clinical dementia rating; Max=maximum; Min=minimum; Mod.=moderate; Sev.=severe; SNP=single-nucleotide polymorphism.
Reproduced from Holtzman DM et al. Alzheimer's disease: the challenge of the second century. Sci Transl Med. 2011;3(77):77sr1. With permission from the American Association for the Advancement of Science.
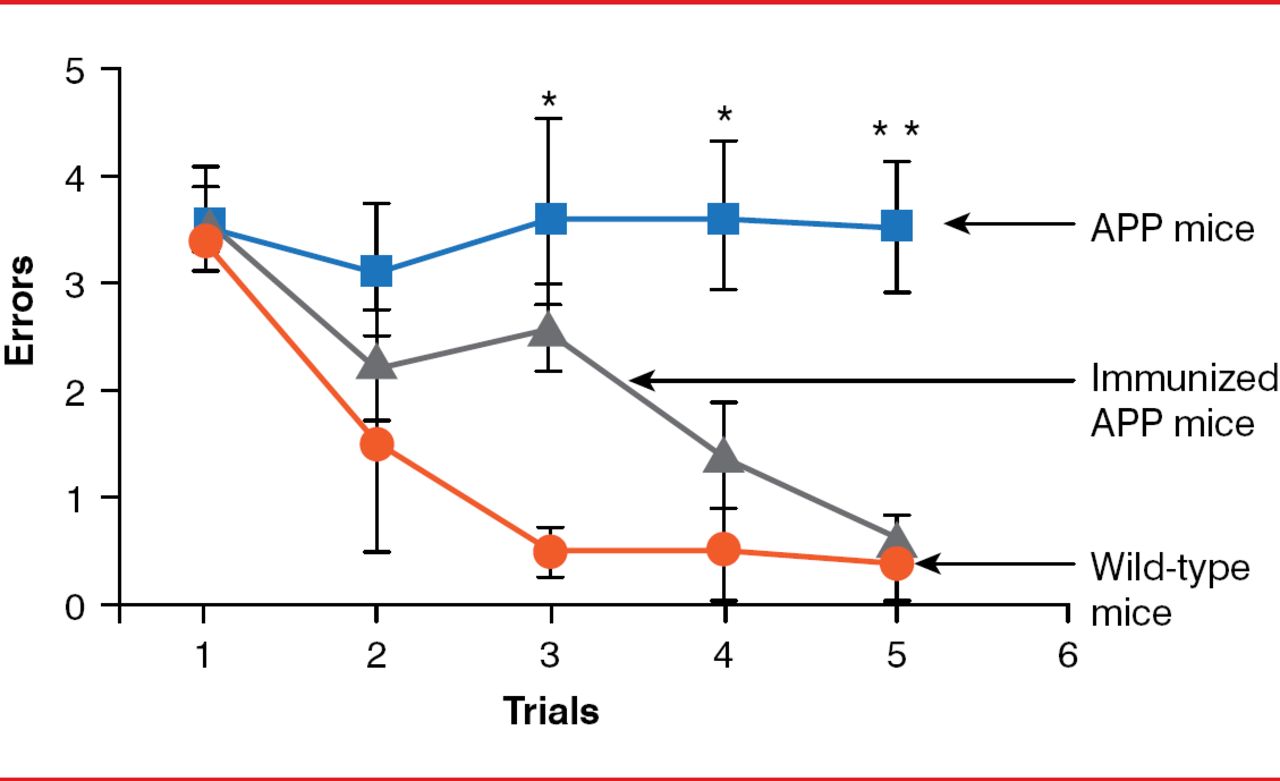
Mice expressing the transgenic β-amyloid precursor protein (APPβ), which increases production of the 42-amino acid form of the peptide (Aβ 42), predominantly found in the amyloid plaques of AD, were first used to study AD treatments. Besides Aβ deposits, these mice develop neuritic plaques, gliosis, and cognitive abnormalities similar to AD in humans. However, mice show no strong signs of tauopathy or prominent neuronal cell loss. The first positive results were noted when transgenic mice were immunized with Aβ 42. When mice were immunized at young ages (6 to 8 weeks), they developed very little amyloid plaques. In older animals that received active immunization (11 months of age), progression of amyloid-related neuropathologies was reduced [Schenk D et al. Nature 1999]. The following year, data indicated that APP-transgenic mice immunized with Aβ 42 had improved learning ability (Figure 3) [Janus C et al. Nature 2000].

Effect of Amyloid-β Immunization on Learning Ability of APP-Transgenic Mice
APP=amyloid precursor protein.
Reproduced from Janus C et al. A beta peptide immunization reduces behavioural impairment and plaques in a model of Alzheimer's disease. Nature. 2000;408(6815):979–982. With permission from the Nature Publishing Group.
Subsequent to this, it was reported that peripheral administration of Aβ antibodies reduces amyloid plaques, with the antibodies triggering microglial cells to clear plaques through Fc receptor-mediated phagocytosis and subsequent degradation of peptides [Bard F et al. Nat Med 2000]. One of these antibodies, called bapineuzumab, was humanized and went into clinical trials. Another anti-Aβ antibody, m266, when given in a prevention mode to APP-transgenic mice, also decreased Aβ levels in the brain, and it also went into clinical trials, as solanezumab [DeMattos RB et al. Proc Natl Acad Sci USA 2001].
Several mechanisms for how these antibodies against Aβ work have been proposed. One suggestion is through clearance of amyloid plaques by phagocytosis. Evidence for this mechanism was shown in several studies. One study showed that the direct application of the anti-Aβ antibody 10D5 to the brain surface of APP mice decreased deposited Aβ and increased microglial infiltration [Bacskai BJ et al. Nat Med 2001]. Imaging of Aβ deposits in brains of living mice permits direct observation of clearance of plaques with immunotherapy [Nat Med 2001]. Other suggested mechanisms include a shift in equilibrium from amyloid plaques to Aβ monomers and blockade of soluble Aβ toxicity (analogous to neutralizing the part of Aβ that damages nerve cells) [Brody DL, Holtzman DM. Annu Rev Neurosci 2008].
Trials in humans with passive immunization targeting Aβ were usually initiated in patients with mild to moderate dementia. At this point, many of these patients have large amounts of amyloid deposition and good amounts of tau deposits throughout the neocortex, suggesting that there may be too much brain damage to reverse the changes substantially [Salloway S et al. N Engl J Med 2014].
In 2 recently published Phase 3 trials in patients with mild to moderate AD, bapineuzumab and solanezumab failed to improve cognition or functional ability as measured in the primary endpoints [Doody RS et al. N Engl J Med 2014; Salloway S et al. N Engl J Med 2014]; however, there were some significant effects that were positive on the secondary endpoints in patients with mild dementia (Alzheimer's Disease Assessment Scale-Cognitive score and others) in the solanezumab trial.
Treating patients with moderate dementia with anti-Aβ therapies may be too late. Thus, solanezumab is being tested in patients with mild AD as well as in asymptomatic individuals with gene mutations that cause dominantly inherited AD and in individuals older than 65 years who have amyloid plaques. Newer anti-Aβ therapies that may remove plaques more extensively (eg, crenezumab, gantenerumab) are also in development and being tested in the preclinical and early clinical phases of the disease.
Dr. Holtzman suggested that future trials might need to increase the doses used, despite the potential for brain swelling noted with bapineuzumab, or to use combination therapies that decrease Aβ production, enhance Aβ clearance, and neutralize Aβ toxicity. Immunotherapy specifically designed to block transcellular tau aggregate propagation may be also be a productive treatment strategy. Antitau monoclonal antibodies (eg, HJ8.5) infused into tau-transgenic mice diminished cognitive deficits and reduced the accumulation of insoluble tau protein in the cortex [Yanamandra K et al. Neuron 2013].
Dr. Holtzman concluded that active and passive immunization against Aβ using antibodies with the best therapeutic properties administered in the preclinical stage of AD has the highest likelihood of success for this approach. Targeting tau with an immunotherapeutic agent, though still in its very early stages, also appears to be a promising approach.
- © 2014 MD Conference Express®
Tools









{kind=link}
{kind=link}
{kind=link}
Table of contents
Cited By...
- No citing articles found.