

Summary
This article discusses the mechanisms of salt-sensitive hypertension. The link between salt intake and hypertension is not a recent discovery but, rather, has been known for a half-century on the basis of studies conducted worldwide. Subjects with hypertension can be classified as salt-insensitive and salt-sensitive. It is beginning to be understood that salt sensitivity can be modulated by multiple mechanisms, including the renal sympathetic nervous system, the mineralocorticoid receptor, and aldosterone.
- Hypertensive Disease
- Cardiology & Cardiovascular Medicine
- Hypertensive Disease
Toshiro Fujita, MD, University of Tokyo, Tokyo, Japan, discussed the mechanisms of salt-sensitive hypertension. The link between salt intake and hypertension is not a recent discovery but, rather, has been known for a half-century on the basis of studies conducted worldwide. Subjects with hypertension can be classified as salt-insensitive and salt-sensitive. The former group exhibits a stable blood pressure (BP) with different levels of salt intake (as measured by the urinary excretion of sodium). Subjects with salt-sensitive hypertension display increasing BP with increased salt intake. It is beginning to be understood that salt sensitivity can be modulated by multiple mechanisms, including the renal sympathetic nervous system, the mineralocorticoid receptor, and aldosterone.
Excessive salt intake in the context of elevated aldosterone leads to the activation of the mineralocorticoid receptor and aldosterone-sensitive epithelial sodium channels in distal nephrons. The result is increased sodium reabsorption, which drives hypertension [Shibata S et al. Hypertension 2007] and proteinuria [Nagase M et al. J Am Soc Nephrol 2006]. This phenomenon was illustrated in experiments conducted in Sprague-Dawley rats fed a low-salt or high-salt diet in the background of a continuous infusion of aldosterone. BP increased linearly with the time of treatment only for those with a high-salt diet, whereas control rats and those fed the low-salt diet with aldosterone infusion displayed a similar pattern of near-constant and significantly lower BP [Shibata S et al. Hypertension 2007]. The rats fed the high-fat diet and treated with aldosterone also displayed markedly higher levels of urinary protein. Another study demonstrated proteinuria in obese rats in the presence of excess aldosterone, which was normalized after treatment with a mineralocorticoid receptor blocker [Nagase M et al. J Am Soc Nephrol 2006].
It appears that activation of the mineralocorticoid receptor may promote hypertension, chronic kidney disease [Nagase M et al. Hypertension 2007], and cardiovascular disease [Matsui H et al. Hypertension 2008] not only in an aldosterone-dependent fashion, as discussed above, but also independent of aldosterone. The observation that plasma aldosterone concentration is not always elevated in obese subjects and that treatment with an aldosterone blocker decreases proteinuria in obese hypertensive subjects suggests that activation of the mineralocorticoid receptor can also occur independent of aldosterone. The identity of the other aldosterone activator(s) remains unclear. Known activators of steroid receptors include, however, SRC-1, Ras, mitogen-activated protein kinase, Smad3, protein kinase A, and Ubc9.
Another potential candidate protein is Rac1, a small G protein that is involved in regulation of the actin cytoskeleton, cell migration, and the response to oxidative stress. It is thought that Rac1 may function in concert with aldosterone in the activation of the mineralocorticoid receptor, which preludes salt-sensitive hypertension and renal damage [Shibata S et al. Nature Medicine 2008]. Experiments in salt-sensitive rat strains (Dahl-S) implicated Rac1 involvement at the level of the kidney in mineralocorticoid receptor—dependent hypertension [Shibata S. J Clin Invest 2011]. Cross-transplant experiments involving renal homografts highlighted the importance of the kidneys in the process of hypertension-related damage in Dahl-S rats [Dahl LK, Heien M. Circ Res 1975].
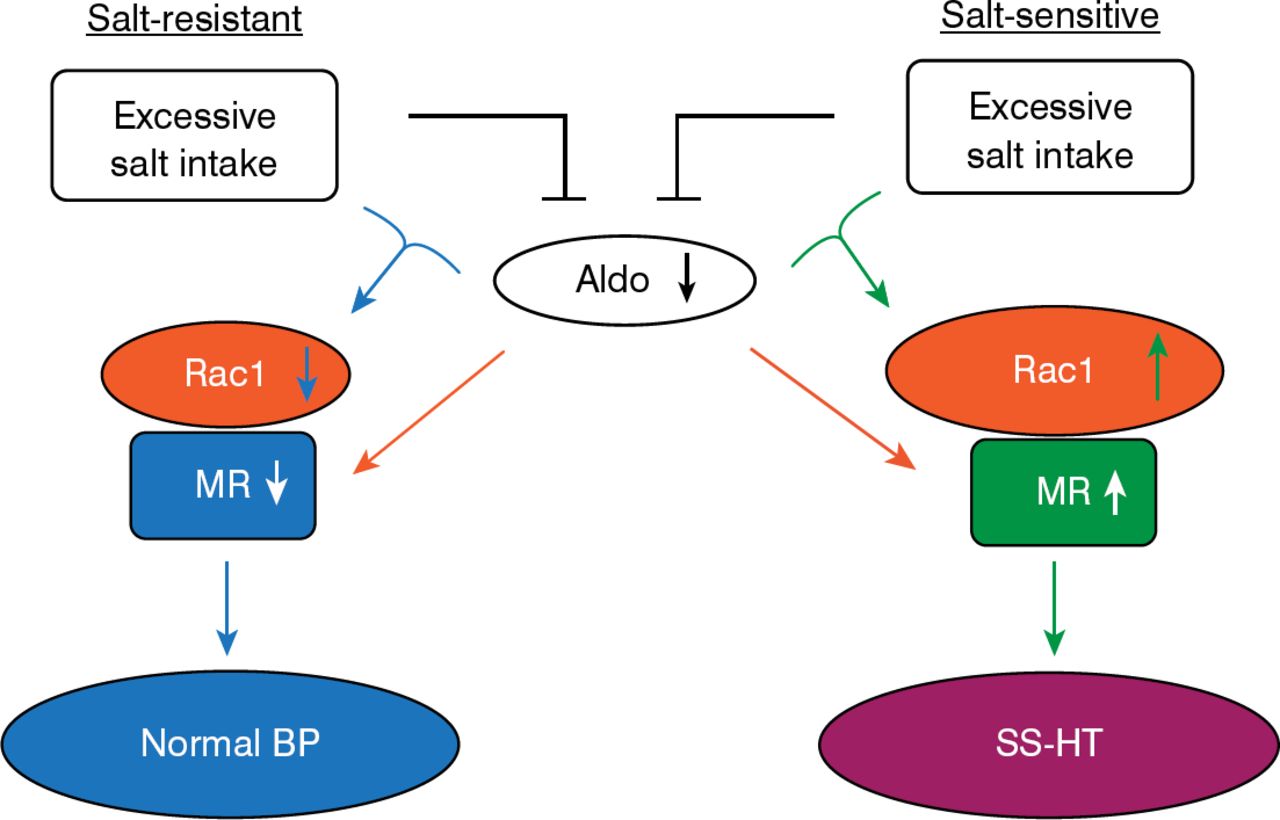
A model to explain these observations is summarized in Figure 1. In the model, downregulation of aldosterone production occurs with excessive salt intake. In salt-resistant organisms, Rac1 and mineralocorticoid receptor are downregulated, resulting in normotension. In salt-sensitive organisms, Rac1 and the mineralocorticoid receptor are upregulated, leading to BP elevation [Shibata S et al. J Clin Invest 2011; Shibata S et al. Nature Medicine 2008].

Renal Rac1 and Salt Sensitivity
Aldo=aldosterone; BP=blood pressure; MR=mineralocorticoid receptor; SS-HT=salt-sensitive hypertensive.
Reproduced with permission from T Fujita, MD.
In addition to aldosterone and the mineralocorticoid receptor, the renal sympathetic nervous system has been implicated in salt sensitivity. Salt loading increases renal sympathetic nervous system activity in salt-sensitive hypertensive rats compared with normotensive rats. In one study, salt-insensitive rats displayed a similar pattern of renal turnover of norepinephrine whether they consumed a low-salt or high-salt diet, whereas salt-sensitive rats had a significantly (p<0.01) more pronounced decline in norepinephrine turnover when consuming a high-salt diet [Fujita T, Sato Y. Hypertension 1992]. In other experiments, the infusion of norepinephrine induced salt-sensitive hypertension in mice, which was associated with the downregulation of renal WNK4 and upregulation of the sodium chloride (NaCl) cotransporter.
The root of this effect may be the increased action of brain reactive oxygen species in the presence of elevated levels of salt (and in obese individuals) [Nagae A et al. Circulation 2009]. Renal sympathetic nervous system overactivity may downregulate the WNK4 protein, which in turn activates the NaCl cotransporter located in the distal convoluted tubules. This activation leads to retention of sodium and, consequently, hypertension [Mu S et al. Nature Medicine 2011].
The glucocorticoid receptor has also been implicated in salt-sensitive hypertension in mice. Normal mice continuously exposed to isoproterenol, a beta adrenergic agonist, developed hypertension when dietary salt levels were elevated. In contrast, glucocorticoid receptor knockout mice displayed no change in mean arterial pressure in the continuous presence of isoproterenol and in high dietary salt concentrations.
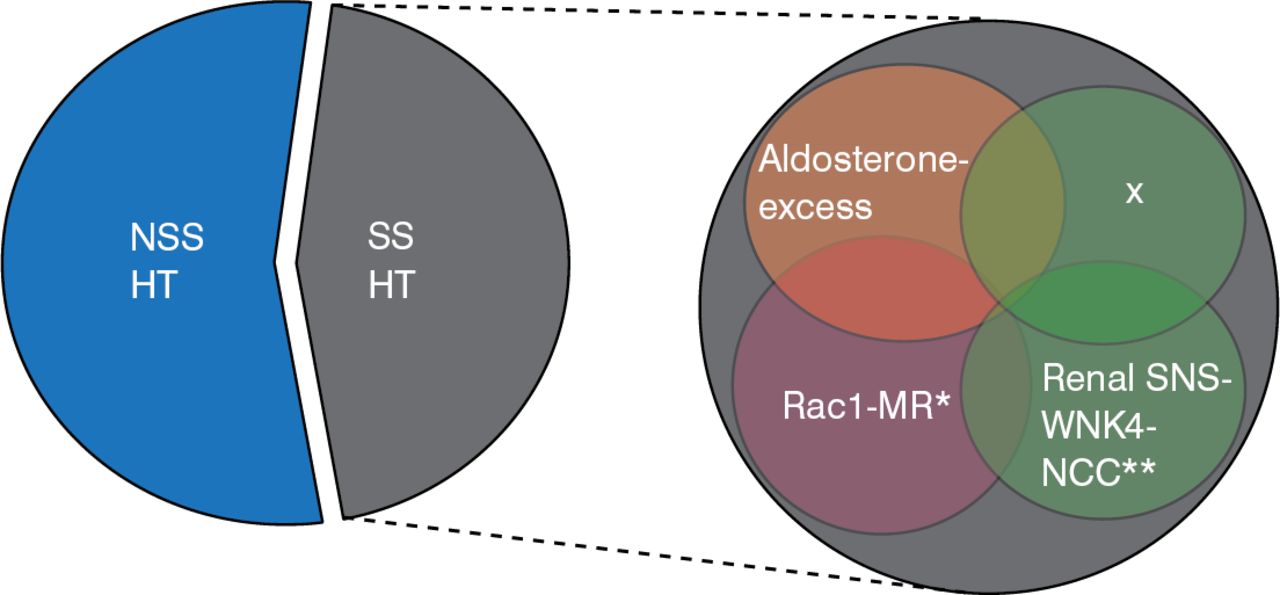
Given the complexity of the mechanisms identified to date for salt-sensitive hypertension, it is likely that other, unidentified factors contribute to this phenomenon in humans (Figure 2).

Complexity of Salt-Sensitive Hypertension in Humans
NSS HT=non-salt-sensitive hypertensive; SS HT=salt-sensitive hypertensive; MR=mineralocorticoid receptor
Reproduced with permission from T Fujita, MD.
*On November 21, 2014, this was changed from Racl-MR to Rac1-MR. **On November 21, 2014, this was changed from SNS-WNK-NCC to SNS-WNK4-NCC.
From an evolutionary perspective, aldosterone may have promoted survival as organisms moved from the sea to the relatively low-salt terrestrial environment by driving sodium homeostasis. As human salt intake began to increase thousands of years ago, plasma aldosterone became suppressed by inhibition of the renin—angiotensin system, which resulted in normal BP. It appears, however, that the modern-day prevalence of high-salt diets and obesity may have exaggerated the role of the Rac1–mineralocorticoid receptor and SNS—WNK4–NCC pathways in BP control, leading to an increasing prevalence of salt-sensitive hypertension. Salt-sensitive hypertension, chronic kidney disease, and cardiovascular disease are examples of what Dr. Fujita termed “diseases of civilization.”
- © 2014 MD Conference Express®
Tools









{kind=link}
{kind=link}
Table of contents
Cited By...
- No citing articles found.