

Summary
Ten years after the identification of the JAK2V617F mutation in most patients with myeloproliferative neoplasms, the understanding of the pathophysiology and genomic landscape of MPNs has evolved. The enhanced understanding and recognition of the impact of symptomatic burden on risk have led to targeted therapies and new treatment paradigms.
- treatment strategies
- algorithms
- myelofibrosis
- pathophysiology
- genomics
- ruxoolitinib
- interferon
New treatment paradigms have emerged from an evolving understanding of the pathogenetic mechanisms and the genomic landscape of the myeloproliferative neoplasms (MPNs). Recognizing the symptom burden is essential for assembling risk-based treatment strategies.
Ruben A. Mesa, MD, Mayo Clinic Cancer Center, Scottsdale, Arizona, USA, reviewed the evolving paradigms of therapeutic goals for MPNs. An estimation of the life-threatening potential of myelofibrosis (MF) and an assessment of the burden of the disease is essential. The risks of vascular events, cytopenias, splenomegaly, transformation of polycythemia vera (PV) and essential thrombocythemia (ET) to MF or acute myelogenous leukemia should all be considered. An important consideration in predicting the threat of the disease is the presence of anemia.
Concern should be even greater if the patient is red cell transfusion-dependent, has a white-cell count > 25 x 109/L cells, or has a platelet count < 100 x 109/L [Passamonti F. Blood. 2012; Barbui T et al. J Clin Oncol. 2011]. Age > 65 years, the presence of significant constitutional symptoms, blasts in the peripheral blood, and certain chromosomal changes are also important predictors. Three or more of these features can indicate that the disease may be life-threatening over the next few years.
Symptomatic burden in MPNs is present in most patients and can compromise quality of life. Symptom management is a prime directive for all MPNs. Quantifying the symptom burden can be achieved through the use of the Myeloproliferative Neoplasm Symptom Assessment Form (MPN-SAF), a comprehensive and reliable instrument that is available in multiple languages to evaluate symptoms associated with all types of MPNs [Scherber R et al. Blood. 2011], and the 10-item MPN-SAF (MPN-10) [Emanuel RM et al. J Clin Oncol. 2012]. The MPN-SAF provides an overall sense of the symptomatic impact of the disease, including symptoms such as fatigue, night sweats, weight loss, fevers, chills, and enlargement of the spleen. Symptomatic burden affects the need for therapy and determining the type of therapy.
The drug landscape for MPNs in 2014 includes cytoreductive agents, single-agent Janus kinase (JAK) inhibitors, combination approaches with a JAK inhibitor, and non-JAK-targeted agents. Successful management of MPN along its disease course may incorporate several of these approaches alone or in sequence, said Dr Mesa. Proposed algorithms for the treatment of ET/PV and MPN-MF rely on calculation of risk and assessment of MPN symptoms.
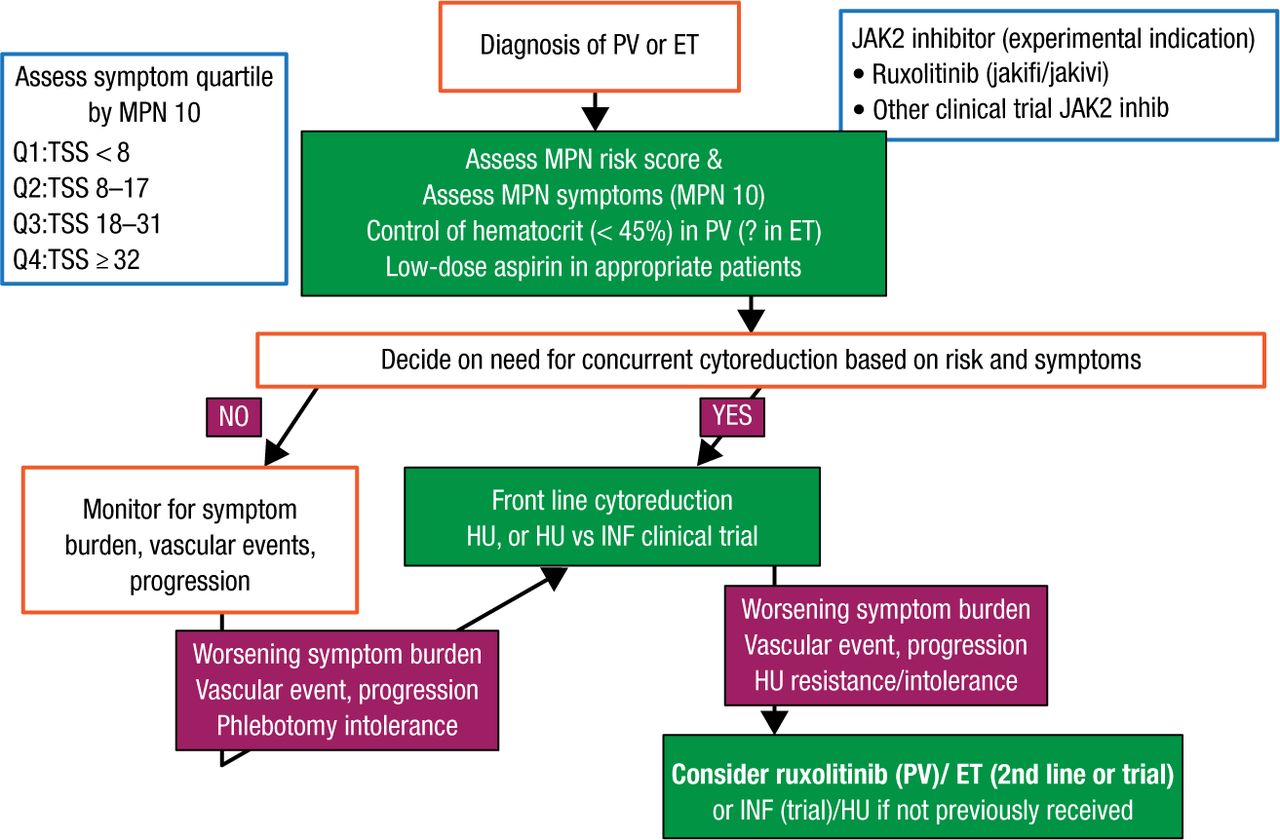
For ET/PV, the focus should be on prevention of thrombohemorrhagic complications and management of symptoms (Figure 1). Assessment should include the MPN risk score and MPN-10 at the time of diagnosis. Stratifying MPN symptom burden by quartile of MPN-10 simplifies objective groupings and allows for objective evaluation of symptomatic response. Hematocrit should be controlled to < 45% in patients with PV and patients should be treated with low-dose aspirin [Barbui T et al. J Clin Oncol. 2011]. Selecting patients with PV/ET for cytoreductive therapy should incorporate the MPN risk score and clinical scenario, such as intolerance of phlebotomy. Hydroxyurea is front-line therapy for both disorders but should be used with caution in younger patients. Anagrelide and interferon are second-line choices in ET and PV, respectively. High-risk patients with PV who have a worsening symptom burden, a vascular event, or cytoreductive resistance despite therapy should be considered for the JAK2 inhibitor ruxolitinib.

Proposed Algorithm of Therapy of ET/PV
ET, essential thrombocythemia; INF, interferon; HU, hydroxyurea; JAK2, Janus kinase 2; MPN, myeloproliferative neoplasms; PV, polycythemia vera; TSS, total symptom score.
Reproduced with permission from RA Mesa, MD.
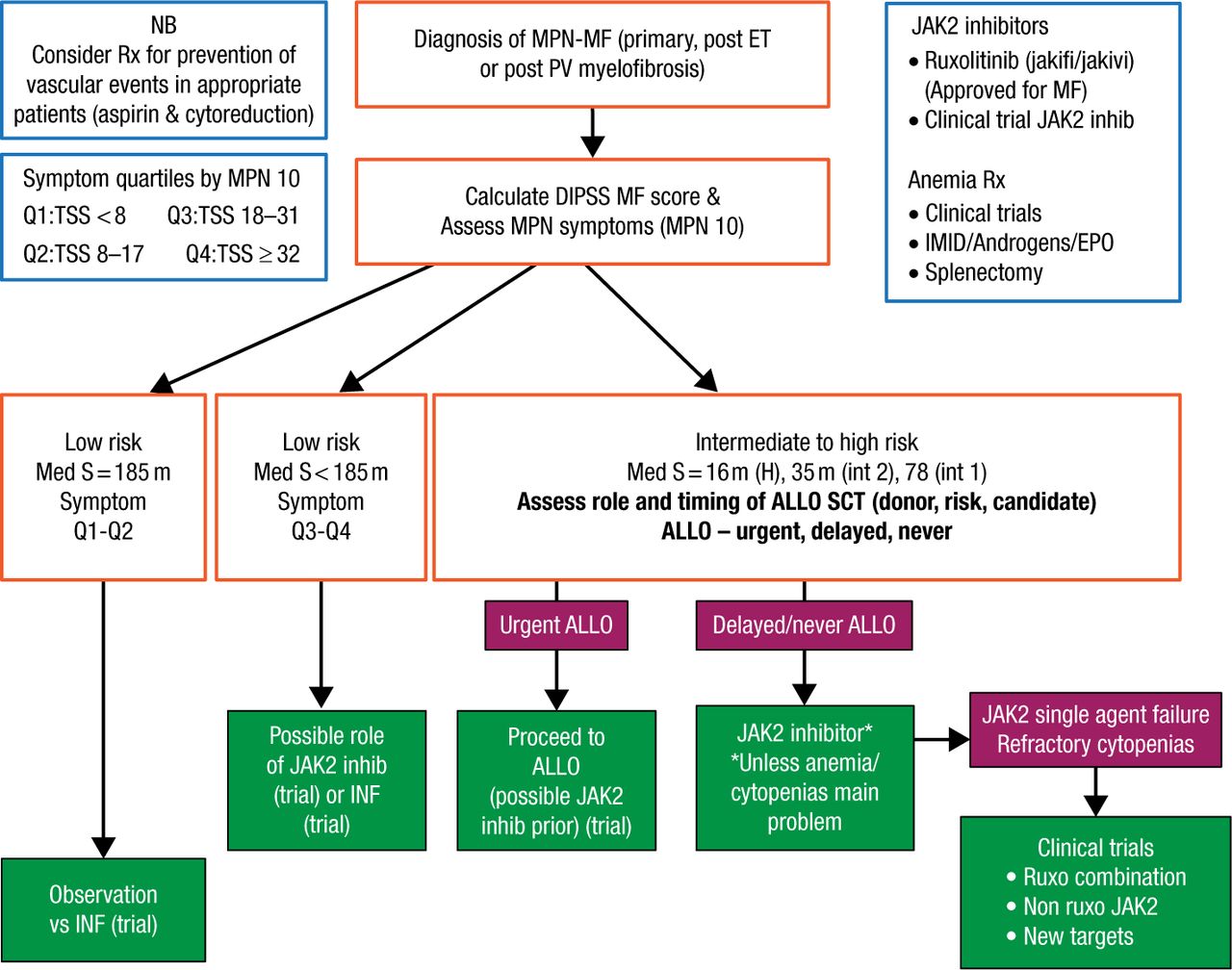
According to Dr Mesa, management of MF should be guided by risk; low-risk patients with low-symptom burdens may be observed or considered for a trial with interferon (Figure 2). Ruxolitinib or interferon may be considered in low-risk patients with a higher symptom burden. Intermediate- to high-risk patients should be immediately assessed for allogeneic hematopoietic stem-cell transplantation (allo SCT) candidacy. For patients deemed not to be allo-SCT candidates, Dr Mesa recommends that ruxolitinib be initiated or that the patient is considered for enrollment into a clinical trial of a JAK2 inhibitor. Patients who do not respond to a JAK2 inhibitor should be considered for trials with other JAK2 inhibitors, ruxolitinib combinations, or non-JAK2 inhibitors.

Proposed Algorithm of Therapy of MPNs/MF
ALLO, allograft; ALLO SCT, allogeneic hematopoietic stem-cell transplantation (allo SCT); DIPSS, Dynamic International Prognostic Scoring System; EPO, erythropoietin; H, high; IFN, interferon; IMID, immunomodulatory drug; int, intermediate; JAK2, Janus kinase 2; MF, myelofibrosis; MPN, myeloproliferative neoplasms; NB, nota bene, Latin for “please note”; PV, polycythemia vera; S, symptoms; TSS, total symptom score.
Reproduced with permission from RA Mesa, MD.
Anthony R. Green, University of Cambridge, Cambridge, United Kingdom, outlined recent advances in understanding the genomic landscape of the MPNs.
According to Mr Green, phenotypic mutations in MPNs affect erythroid and megakaryocyte signaling pathways. Mutations in JAK2 and myeloproliferative leukemia are gain-of-function mutations that result in cytokine-independent growth in cell lines with downstream activation of JAK-STAT signaling. JAK2-unmutated MPNs, which encompass about half of patients with ET or MF, represent distinct subgroups with differences in clinical features at presentation and clinical outcome [Klampfl T et al. N Engl J Med. 2013].
Up to 80% of patients with MPNs with nonmutated JAK2 were found to have recurrent somatic mutations in the gene calreticulin (CALR) [Klampfl T et al. N Engl J Med. 2013; Nangalia J et al. N Engl J Med. 2013]. CALR mutations are all insertions or deletions in the DNA sequence of exon 9.
CALR is a highly conserved luminal endoplasmic reticulum chaperone protein that ensures the proper folding of newly synthesized glycoproteins and has also been implicated in phagocytosis, calcium homeostasis, immunogenic cell death, proliferation, and apoptosis. CALR mutations are acquired at the level of the hematopoietic stem cell, and clonal characterization of MPN samples has shown that mutated CALR is present in the earliest clone, consistent with it being an initiating event in MPNs. In ET, patients with a CALR mutation have an increased risk of transformation to MF. In MF, patients with a CALR mutation have improved survival compared with JAK2-mutated or triple negative patients [Rumi E et al. Blood. 2014], but survival might depend on the type of CALR mutation [Tefferi A et al. Blood. 2014].
JAK2, MPL, and CALR mutations provide a genetic marker for 99% of PV and 85% of ET and MF, but other genes involved in DNA methylation or chromatin structure are also mutated in MPNs. Mutations in epigenetic regulators (TET2, IDH1/2, DNMT3A, EZH2, and ASXL1) are present in different MPNs and are especially common in MF and/or blast phase. These mutations influence DNA methylation and histone modification. Mutations affecting DNA methylation may give rise to hematopoietic stem and progenitor cells’ (HSPC) advantage needed for clonal expansion. The hematologic consequences of epigenetic mutations are heterogeneous and include myelodysplasia-like cytopenias and HSPC reduction with ASXL1 knockout and an increase in platelets and white blood cells and HSPC expansion with EZH2 knockout.
In MPNs, a high number of mutated genes predict poorer survival and a greater risk of transformation [Lundberg P et al. Blood. 2014]. In MF, the presence of mutations in any one of ASXL1, EZH2, SRSF2, and IDH1/2 is associated with poorer overall survival and increased leukemic transformation [Vannucchi AM et al. Leukemia. 2013; Guglielmelli P et al. Leukemia. 2014]. The number of these “high molecular risk” mutations is inversely correlated with median survival in MF.
The order of mutation is important in influencing stem and progenitor cell behavior, said Mr Green, as a TET2 mutation before JAK2 mutation prevents JAK2 V617F from upregulating a proliferative transcriptional program.
- © 2014 SAGE Publications
Tools









{kind=link}
{kind=link}
Table of contents
Cited By...
- No citing articles found.