

Summary
This study identifies a potential mechanism of action for lenalidomide in del(5q) myelodysplastic syndrome and identifies a single nonconserved amino acid in mice and humans that determines lenalidomide susceptibility. The results may facilitate the development of improved preclinical models and reinforces the importance of haploinsufficient genes in the pathogenesis and treatment of disease.
- del(5q)
- mouse model
- human cell lines
- proteomics
- ubiquitination
- degradation
- lenalidomide
Lenalidomide is a highly effective treatment for multiple myeloma (MM) and myelodysplastic syndrome (MDS) with deletion of chromosome 5q (del[5q]). In MM, the activity of lenalidomide is thought to be the result of activation of the cereblon (CRBN)-CRL4 E3 ubiquitin ligase to ubiquitinate the transcription factors IKZF1 and IKZF3 [Krönke J et al. Science. 2014]. Emma C. Fink an MD/PhD student at Brigham and Women’s Hospital, Boston, Massachusetts, USA, reported the results of a study [Fink EC et al. ASH 2014 (abstr 4)] indicating that in MDS with del(5q), lenalidomide induces the ubiquitination of casein kinase 1A1 (CSNK1A1) by CRBN-CRL4 and its subsequent degradation by the proteasome. Haploinsufficiency for CSNK1A1 sensitizes del(5q) to lenalidomide treatment and results in p53-dependent killing.
The investigators used global proteomics profiling in the myeloid cell line KG-1 to identify CSNK1A1 as a novel and direct target of lenalidomide. CSNK1A1 is haploinsufficient in del(5q) MDS and is a negative regulator of p53 and β-catenin [Schneider RK et al. Cancer Cell. 2014]. Lenalidomide treatment increased ubiquitination and decreased protein levels of CSNK1A1.
Additional analyses showed that lenalidomide treatment resulted in a dose-dependent decrease in CSNK1A1 protein levels in multiple human cell lines without altering CSNK1A1 mRNA levels. In addition, the investigators found that CSNK1A1 associates with CRBN-CRL4 only in the presence of lenalidomide, suggesting that lenalidomide induces the recruitment of CSNK1A1 to CRBN-CRL4. When mixed in vitro, CSNK1A1 is ubiquitinated by CRBN-CRL4. Ubiquitinated CSNK1A1 is then subsequently degraded by the proteasome leading to low protein levels.
The investigators then analyzed the effects of CSNK1A1 haploinsufficiency on lenalidomide sensitivity in a genetically defined Csnk1a1 conditional knockout mouse model. They found that mouse cells do not respond to lenalidomide. Expression of human CRBN rendered the cells sensitive to lenalidomide, suggesting that sequence differences between mouse and human CRBN, the direct binding partner of lenalidomide, explained this species-specific response. A single amino acid change in mouse CRBN (isoleucine to valine at position 391; I391V) was identified that restored lenalidomide response in mouse CRBN.
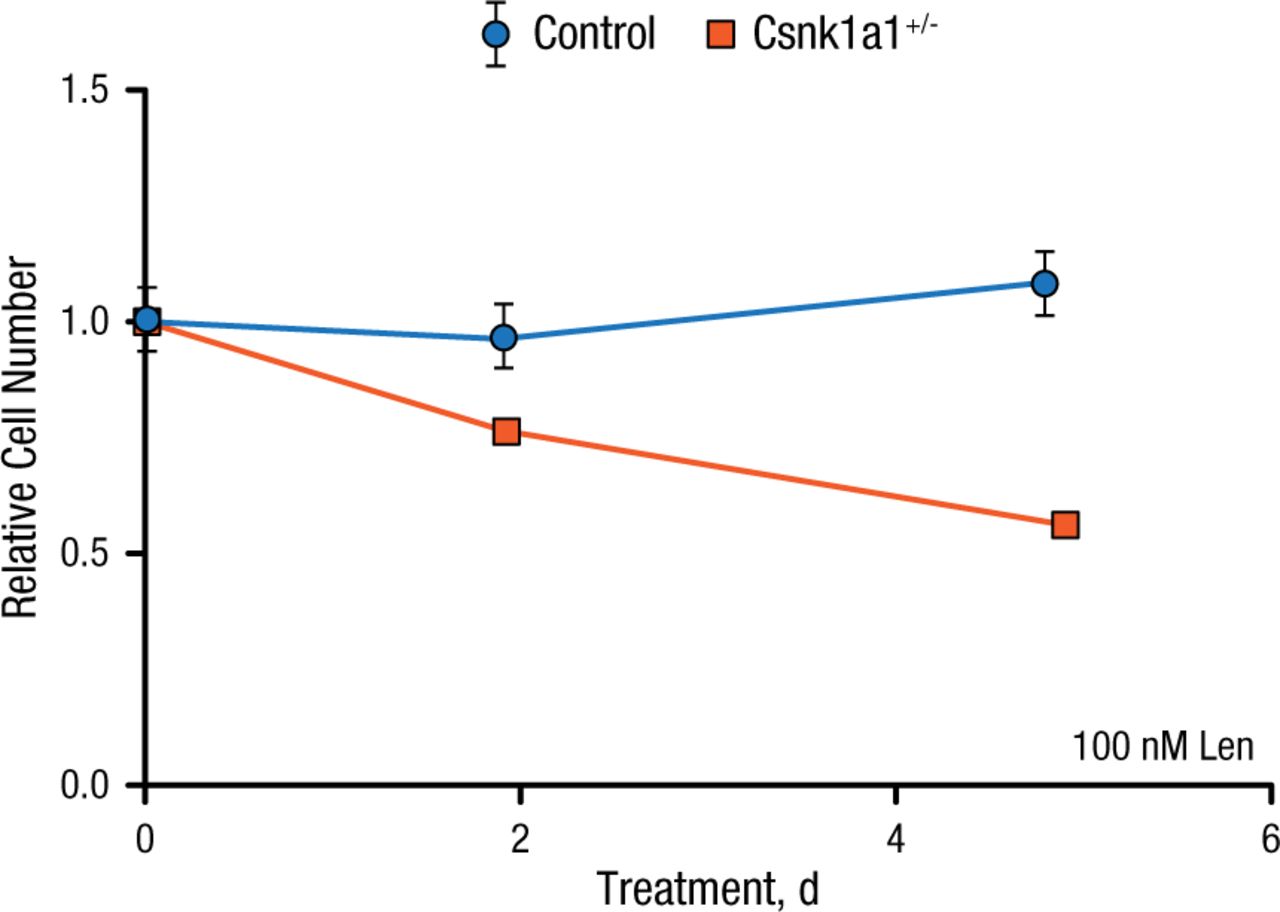
After identifying a method to make mouse cells sensitive to lenalidomide, the investigators returned to the Csnk1a1 conditional knockout mouse model. Bone marrow cells from control mice or mice heterozygous for Csnk1a1 were transduced with the mouse CRBN I391V allele then treated with lenalidomide. Unlike controls, which did not respond to lenalidomide, Csnk1a1+/– cells depleted almost 50% over 5 days of lenalidomide treatment. These results demonstrate that hematopoietic Csnk1a1+/– cells are more sensitive to lenalidomide than wild type cells with 2 copies of the gene (Figure 1).

Csnk1a1+/– Cells Are More Sensitive to Lenalidomide
Csnk1a1, casein kinase 1A1; Len, lenalidomide.
Reproduced with permission from EC Fink, MD.
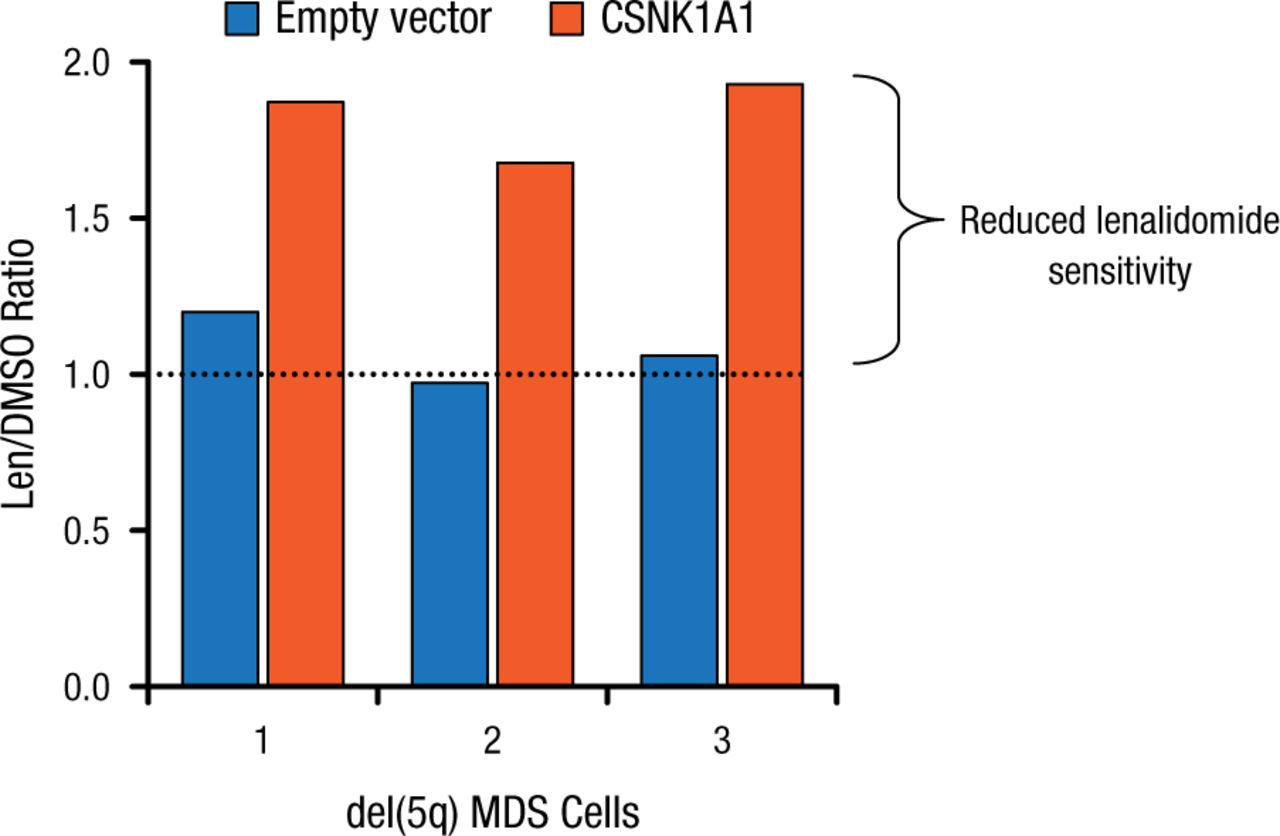
Lenalidomide treatment led to the induction of the p53 target p21 in Csnk1a1+/– cells, suggesting that the activation of the p53 pathway is involved in lenalidomide’s downstream mechanism. Deletion of a single allele of p53 completely rescued the lenalidomide sensitivity of Csnk1a1+/– cells, implying a p53 killing mechanism. Further, CSNK1A1 overexpression reduces lenalidomide sensitivity in del(5q) patient samples (Figure 2).

Effect of Overexpression of CSNK1A1 on Lenalidomide Sensitivity of del(5q)
CSNK1A1, casein kinase 1A1; del(5q), deletion of chromosome 5q; DMSO, dimethyl sulfoxide; len, lenalidomide; MDS, myelodysplastic syndrome.
Reproduced with permission from EC Fink, MD.
To conclude, Dr Fink noted that another study [Macbeth et al. ASH 2014 (abstr 3606)] showed similar results regarding lenalidomide inducing ubiquitination of CSNK1A1 by the CRBN-CRL4 and its subsequent degradation.
- © 2014 SAGE Publications
Tools









{kind=link}
{kind=link}
Table of contents
Cited By...
- No citing articles found.