

Summary
New research findings can now explain the complete pathway for the regulation of thrombopoietin production, which regulates the production of platelets. Platelets lacking sialic acid due to aging are removed by the Ashwell-Morell receptor. The signaling pathways Janus kinase 2 and signal transducer and activator of transcription 3 then stimulates the production of thrombopoietin within the hepatocyte nucleus.
- Ashwell-Morell receptor
- desialyted platelets
- human hepatoma cells
- in vitro
The Ashwell-Morell receptor (AMR) on the hepatocyte binds and removes desialylated platelets as they circulate and age in the blood. After binding to the AMR, the desialylated platelets induce hepatic expression of thrombopoietin (TPO) mRNA, thereby regulating platelet production. Renata Grozovsky, PhD, Brigham and Women’s Hospital, Boston, Massachusetts, USA, presented data from a study [Grozovsky R et al. Nat Medicine. 2015] showing the TPO expression by way of the AMR is further regulated by Janus kinase 2 (JAK2) and signal transducer and activator of transcription 3 (STAT3) protein signaling in vivo and in vitro.
In 1994, 4 studies identified ligands that significantly increased platelet levels [Bartley TD et al. Cell. 1994; de Sauvage FJ et al. Nature. 1994; Kuter DJ et al. Proc Natl Acad Sci USA. 1994; Lok S et al. Nature. 1994]. The substance was subsequently identified as TPO, a glycoprotein hormone produced by the liver and kidney that in humans is encoded by the TPO gene. TPO regulates platelet production, supporting the survival, proliferation, and differentiation of platelet precursors and bone marrow megakaryocytes. The complete understanding of this pathway remained elusive until Dr Grozovsky’s research.
In the in vitro treatment, desialylated platelets were shown to stimulate TPO production in human hepatoma cells (HepG2). The data indicate that following platelet desialylation, platelet ingestion by the AMR, TPO mRNA and the TPO protein significantly increase within 6 hours (P < .01) [Grozovsky R et al. Nat Medicine 2015].
Desialylated human platelets were also shown to activate JAK2-STAT3 signaling in HepG2 cells (Figure 1). Phospho-STAT3 was translocated into the nucleus and TPO protein expression increased as well. Both of the latter were prevented by pharmacological JAK inhibition.

Desialylated Human Platelets Stimulate JAK2 and STAT3 Phosphorylation in HepG2 Cells
JACK2, Janus kinase 2; STAT3, activator of transcription 3.
*P < .05, **P < .01
Reproduced with permission from R Grozovsky, PhD.
In the in vivo treatment, AMR-deficient mice had increased platelet count, survival, and loss of sialic acid compared with wild type mice showing that removal of desialylated platelets by the AMR regulated in vivo platelet survival.
In livers isolated from Asgr2-null mice, TPO mRNA expression decreased by 40%, while TPO mRNA expression in livers from mice lacking sialyltransferase (St3gal4-null) was increased by 30%, compared with wild type mice. In St3gal4-null mice, desialylated platelet clearance is increased and specifically mediated by the AMR. This indicates that desialylated platelet uptake by the AMR regulates liver TPO levels.
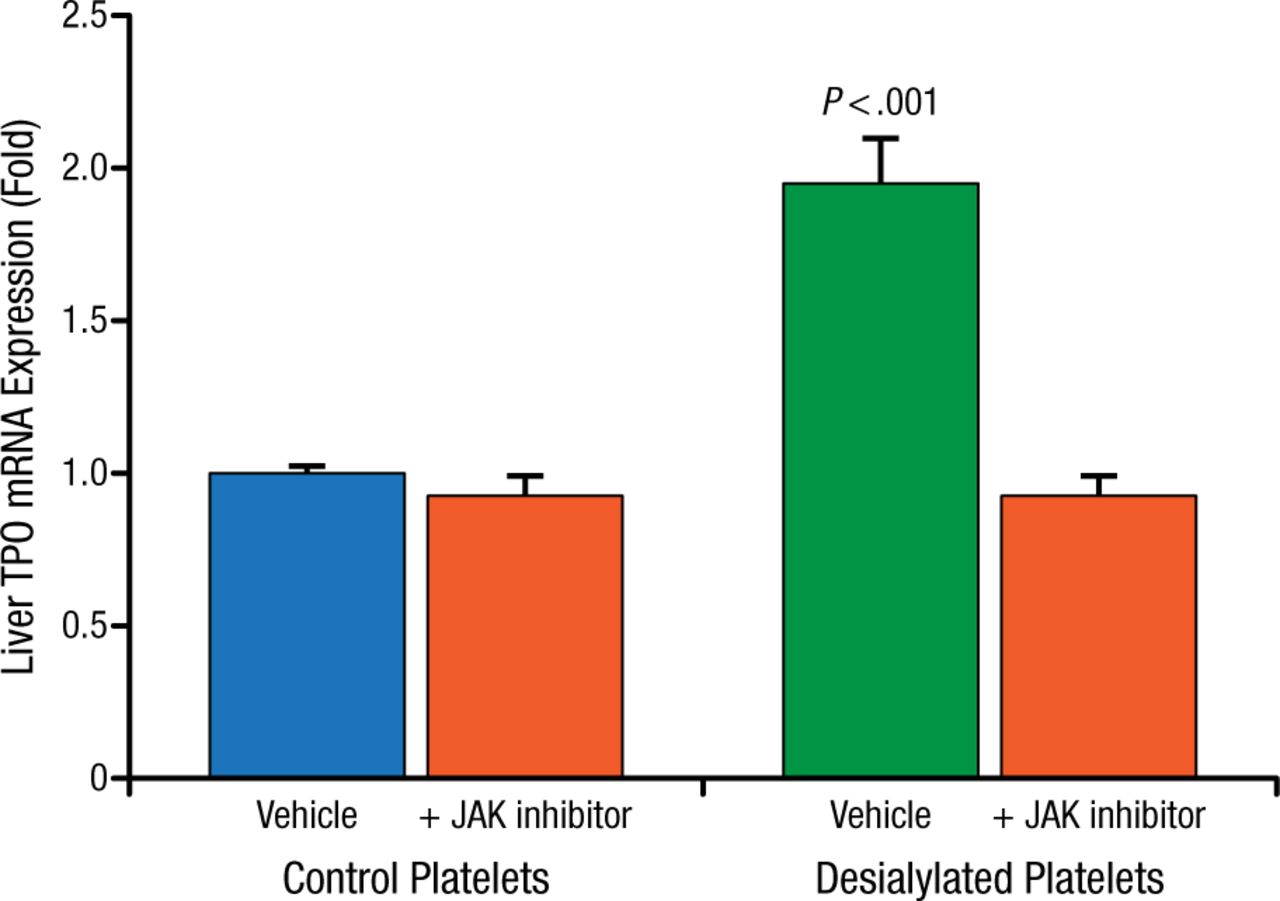
When desialylated platelets isolated from St3gal4-null or Asgr2-null mice are infused into wild type mice, hepatic TPO mRNA levels increase as early as 12 hours after infusion, while desialylated platelets infused into Asgr2-null mice have no effect on TPO mRNA synthesis. Further, treatment with JAK inhibitors block the desialylated platelet stimulated TPO mRNA increase in wild type mice (Figure 2).

JAK Inhibitors Block TPO mRNA Increase in Wild Type Mice
JAK, Janus kinase; TPO, thrombopoietin.
Reproduced with permission from R Grozovsky, PhD.
The accumulated data show that circulating desialylated platelets by way of the AMR and JAK2-STAT3 signaling stimulated TPO production, providing a physiological feedback mechanism to regulate plasma TPO levels and platelet production in vivo and in vitro. The complete understanding of this feedback mechanism illuminates the pathophysiology of platelet diseases, such as thrombocythemia and immune thrombocytopenia. This feedback has clinical implications, however, as the administration of JAK1/2 inhibitors can cause thrombocytopenia in some groups of patients.
- © 2014 SAGE Publications
Tools









{kind=link}
{kind=link}
Table of contents
Cited By...
- No citing articles found.