

Summary
Genomic analysis and molecular profiling identified mutations and translocations in ER+ breast cancer that are targets for genome-directed therapeutics. ESR1 mutations and translocations contribute to endocrine therapy resistance. Novel pharmacologic approaches have successfully treated ESR1 Y537 mutation-driven resistance, mutant HER2 tumors, and wild-type RB and TP53 tumor suppressor function in preclinical and clinical work.
- genome-directed therapeutics
- estrogen receptor-positive
- endocrine therapy
- resistance
The development of genome-directed therapeutics (GDT) for estrogen receptor (ER)-positive breast cancer (BC) requires a rational approach to sort through the volume of complex genomic data that are now available to identify mechanisms and possible targets, stated Matthew J. Ellis, MD, PhD, Baylor College of Medicine, Houston, Texas, USA. Molecular profiling and genomics have identified point mutations, gene copy aberrations, and structural abnormalities, including translocations and small and large deletions, which cause BC and help to explain resistance to endocrine therapy.
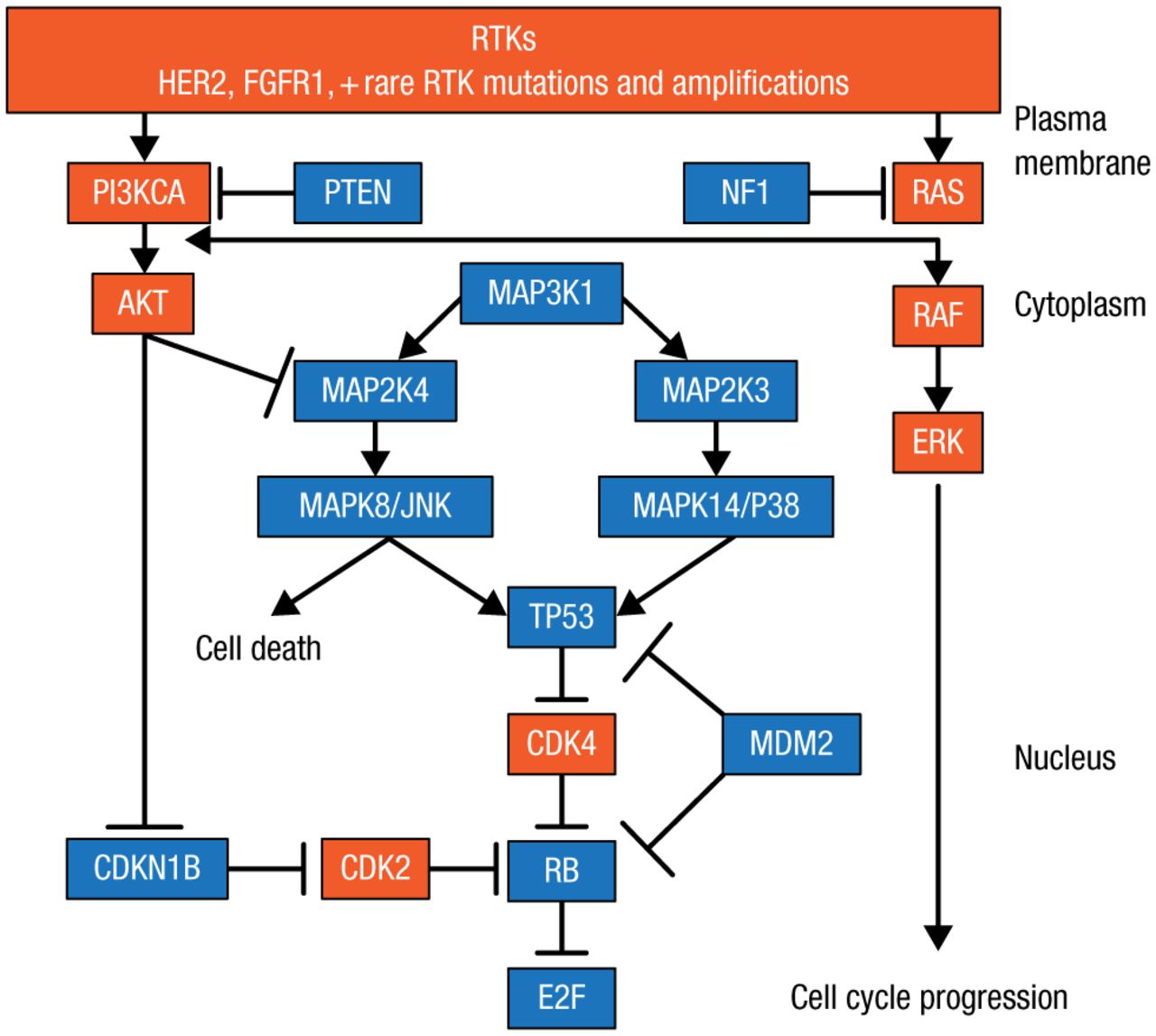
Dr Ellis and colleagues identified significantly mutated genes (SMGs) in tumor biopsies from patients in 2 studies of neoadjuvant aromatase inhibitors (AIs), which explain why some patients respond and others are resistant to AI therapy [Ellis MJ et al. Nature. 2012]. Such lists of SMGs can be used to identify low-frequency and common mutations to draw functional and therapeutic maps to identify new therapeutic targets (Figure 1). A number of nodes are being targeted, including the receptor tyrosine kinase (RTK) mutations, human epidermal growth factor receptor 2 (HER2) mutations, and the PIK3CA mutation in the phosphatidylinositol-3-kinase (PI3K) pathway. The RAS/RAF/MAPK pathway has a low frequency in BC, but it may be important in regard to prognosis, noted Dr Ellis, and luminal-type BC is predominantly TP53 wild-type (WT) mutations, which suggests some important therapeutic opportunities. Dr Ellis therefore focused on the 6 principle nodes that are being targeted: estrogen receptor 1 (ESR1), RTK, RAS, TP53, RB, and PI3K.

Functional and Therapeutic Map of Gene Mutations to Guide Drug Development
HER2, human epidermal growth factor receptor 2; PI3K, phosphatidylinositol-3-kinase; RTK, receptor tyrosine kinase.
Adapted from Cancer Discovery, Copyright 2013;3:27–34, Ellis MJ et al. The Genomic Landscape of Breast Cancer as a Therapeutic Roadmap. With permission from AACR.
Important mechanisms for resistance to endocrine therapy are mutations and translocations in ESR1, which were identified through patient-derived xenografts (PDX) of ER-positive BC [Li S et al. Cell Rep. 2013]. In patients with advanced BC resistant to endocrine therapy, tumor growth was found to be both independent of and dependent on estrogen supplementation. In some cases, estrogen actually delayed tumor growth, and regression was induced in established tumors, supporting the use of estradiol as therapy for advanced breast cancer [Ellis MJ. JAMA. 2009]. Importantly, 4 of the 6 genomes had aberrations in the estrogen receptor, including amplification, translocation, and Y537S and E380Q mutations. Point mutations in the ESR1 ligand-binding domain result in resistance to AI [Toy W et al. Nat Genet. 2013].
Selective estrogen receptor degrading (SERD) agents may be useful to address resistance driven by ESR1 Y537S mutations. Preclinical work has shown that ARN-810, an oral SERD, reduced the tumor volume in a mutant mouse xenograft model of this mutation compared with fulvestrant and vehicle, and a phase 1 dose-finding study of the treatment is currently underway [Bardia A et al. SABCS 2014 (poster P1-13-01)]. ESR1 mutations are present in about 1.9% of primary BC, so these mutations may be important to consider even in early-stage disease [Griffith OL et al. SABCS 2014 (abstr S1-02)].
For the RB gene without inactivating mutations, restoration of its suppressed activity by inhibiting RB-inactivating phosphorylations with a CD4/6 inhibitor may be therapeutic. One study showed that the CD4/6 inhibitor palbociclib, compared with vehicle, had a cytostatic effect in WHIM18-endocrine refractory BC that harbored an endocrine therapy resistance-associated ESR1 translocation.
Regarding the RTK node, about 2% of BC cases have HER2 mutations [Ellis MJ, Perou CM. Cancer Discov. 2013]. The most common mutation, L755S, is resistant to lapatinib but has been shown to respond to neratinib. However, acquired resistance to neratinib is an issue that has led to consideration of possible combination therapies. Dr Ellis believes that combining endocrine therapy and neratinib is the next step.
The RAS node is a somewhat forgotten pathway in BC, according to Dr Ellis. In this setting, although inactivating mutations in the RAS negative regulator NF1 are found in only about 1% of BC cases, it seems to be a lethal mutation. Exploiting the clinical trial database for neurofibromatosis, a disease driven by truncating NF1 mutations, may lead to therapies to target loss of function in BC. One approach may be targeting constitutive RAS downstream with a RAF and MEK combination [Long GV et al. N Engl J Med 2014].
Restoring p53 function may lead to tumor suppression and control of tumor growth. One target is MDM2, an E3 ligase that degrades the p53 protein. MI-773, an MDM2 inhibitor, increased levels of MDM2, p53, and downstream targets of p53 in preclinical work [Wang S et al. Cancer Res. 2014]. Further work showed that ESR1 YAP (Yes-associated protein) translocation that did not respond to endocrine therapy responded to MI-773, with tumor mean volume remaining constant. When MDM2 was amplified, there was also a response to MI-773, including tumor regression. In collaborative work, Dr Ellis and colleagues are finding that in TP53 WT luminal tumors, very low levels of TP53 may be due to upregulation of other E3 ligases and the genes that regulate E3 ligases. Thus, he believes there are other possible targets in the TP53 degradation pathway beyond MDM2.
The PI3K pathway is an important therapeutic target because of the results of the BOLERO-2 study [Baselga J et al. N Engl J Med. 2012]. However, a sensitivity/resistance algorithm was not identified by tumor sequencing of this study, probably because most of the samples came from the primary, not the metastatic, sample. It has become abundantly clear that tumors that have progressed through multiple lines of therapy have a very different genomic repertoire. Many new agents targeting PI3K are being investigated.
Among the approaches to treat metastatic BC or primary ER-positive BC being investigated is targeting > 2 nodes. Trials are underway that are targeting ESR1, PI3K, and RB simultaneously. However, Dr Ellis stated this approach will work only in genomically defined subgroups of patients, and such triplets must be used in a rational genome-directed manner. Genomic analysis of metastatic disease is an essential prerequisite for logical clinical trial development. Neoadjuvant endocrine therapy trials may lead to genome-driven combination studies in the early disease setting.
- © 2014 SAGE Publications
Tools









{kind=link}
Table of contents
Cited By...
- No citing articles found.