

Summary
The two forms of large-vessel (LV) vasculitis—giant cell arteritis and Takayasu arteritis—pose a clinical challenge because they are relatively uncommon, complex to diagnose, and have few treatment options. This article reviews the epidemiology and management of patients with LV vasculitis and highlighted unmet needs with respect to diagnosis and treatment choices.
- Vasculitis
- Rheumatology
- Vasculitis
The two forms of large-vessel (LV) vasculitis—giant cell arteritis (GCA) and Takayasu arteritis (TAK)—pose a clinical challenge because they are relatively uncommon, complex to diagnose, and have few treatment options. This session reviewed the epidemiology and management of patients with LV vasculitis and highlighted unmet needs with respect to diagnosis and treatment choices.
GIANT CELL ARTERITIS: BEYOND STEROID THERAPY
Bhaskar Dasgupta, MD, MRCP, Southend University Hospital, Essex, United Kingdom, discussed the management of patients with GCA. Temporal artery biopsy is the gold standard for diagnosis and should be performed on all patients with suspected cranial GCA, Prof. Dasgupta said. Histology is not only diagnostic but can also be prognostic, since the degree of intimal hyperplasia predicts subsequent ischemic complications [Makkuni D et al. Rheumatology (Oxford) 2008]. Temporal artery ultrasound is also valuable for diagnosis, with or without ultrasonography of the axillary artery. The presence of the “halo sign” has high diagnostic accuracy [Karassa FB et al. Ann Intern Med 2005]; also, unlike biopsy, ultrasound allows the entire artery to be assessed. The Temporal Artery Biopsy Versus Ultrasound in Diagnosis of GCA [TABUL; NCT00974883] study is now underway to compare biopsy and ultrasound with regard to diagnostic accuracy and cost-effectiveness. Results are expected in 2014.
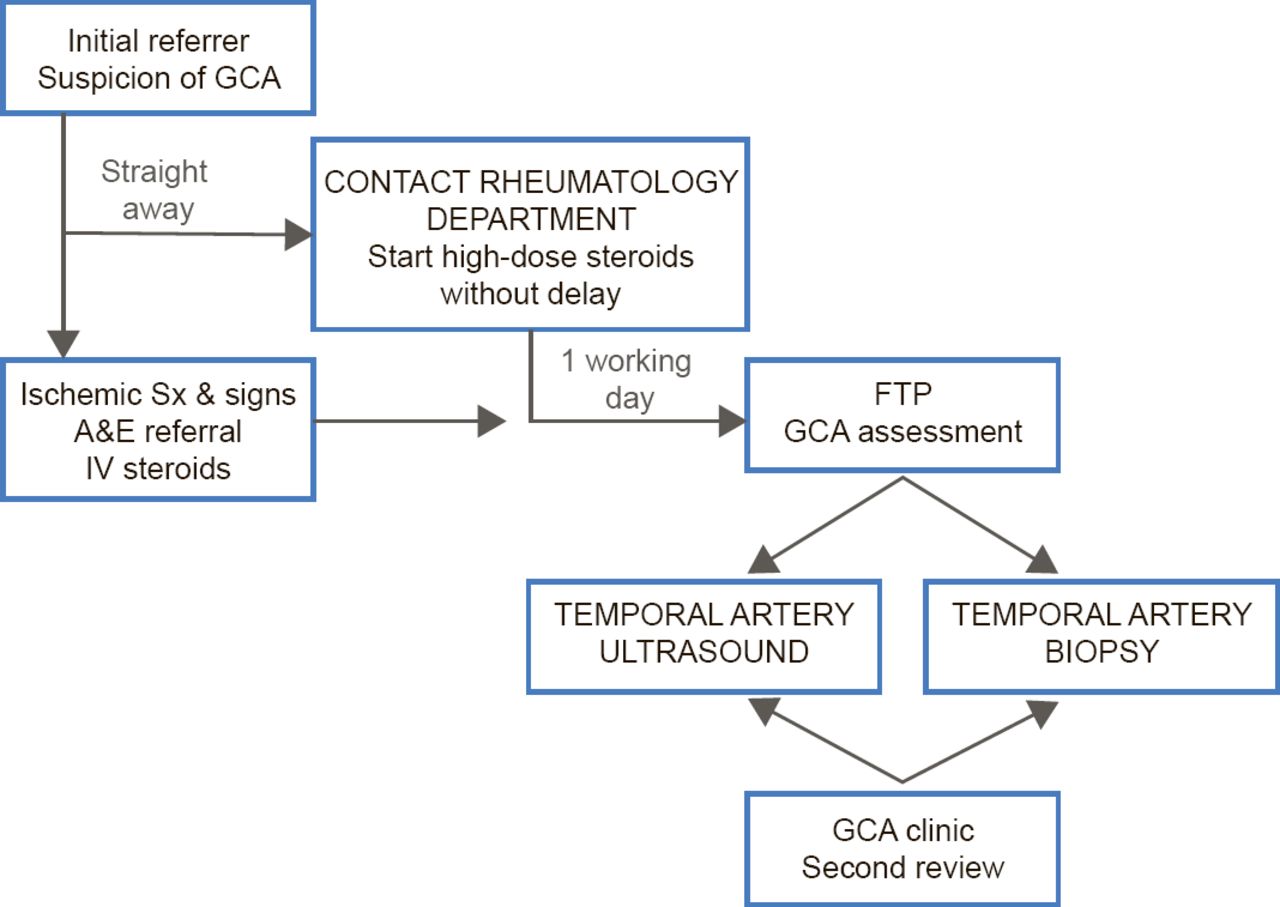
A major manifestation of cranial GCA is visual loss, and Prof. Dasgupta suggested that GCA should be managed using a “fast-track pathway” (FTP) analogous to that used in patients with myocardial infarction or stroke. GCA is a critically ischemic disease, he said, yet the average time from symptom onset to diagnosis is 35 days [Ezeonyegi AN et al. Clin Rheumatol 2011]. To expedite treatment, Southend University Hospital has implemented an FTP (Figure 1) in which patients with suspected GCA are immediately started on high-dose steroids and undergo temporal artery ultrasound and biopsy within 7 days. This protocol has reduced the average time to diagnosis and dramatically improve visual outcomes [Patil P et al. ACR 2013 (abstr 1928)]. Furthermore, the pathway is cost-effective and offers an incremental gain in quality-adjusted life years [Achilleos K et al. ACR 2013 (abstr 2667)].

Fast-Track Pathway for Suspected GCA
A&E=accident and emergency; FTP=fast-track pathway; GCA=giant cell arteritis;
IV=intravenous.
Adapted from Patil P et al. ACR 2013 (Abst 1928).
Long-term steroids are the mainstay of therapy for GCA and, while effective in most patients, they are associated with adverse events such as diabetes and fractures. Better treatments are therefore urgently needed. Disease-modifying agents such as methotrexate have limited efficacy in GCA; leflunomide is effective and well-tolerated but has not been evaluated in a randomized controlled trial (RCT); and biologics to date have been disappointing. Interleukin-6 is a key player in the pathogenesis of GCA and tocilizumab has shown impressive efficacy in refractory patients. An RCT, the Giant Cell Arteritis Clinical Research Study [GiACTA; NCT01791153], is now underway comparing tocilizumab with prednisolone.
LARGE VESSEL VASCULITIS IN GIANT CELL ARTERITIS
Kenneth Warrington, MD, Mayo Clinic College of Medicine, Rochester, Minnesota, USA, reviewed LV vasculitis in patients with GCA. GCA is an extensive vasculopathy that can involve arteries of the upper and lower extremities as well as the aorta, he noted.
The prevalence of upper extremity LV disease among patient with GCA is ∼80% and usually involves the subclavian arteries [Blockmans D et al. Arthritis Rheum 2006]. Lower extremity LV disease is less prevalent, at ∼37%, and usually involves the superficial femoral and popliteal arteries. In both scenarios, bilateral disease is common and symptoms are rare. Claudication of the arm and leg is reported by 4% to 6% and 1% to 20% of those with upper and lower extremity disease, respectively [Schmidt WA et al. Rheumatology (Oxford) 2008; Nuenninghoff DM et al. Arthritis Rheum 2003; Czihal M et al. J Rheumatol 2012].
Compared with cranial artery GCA, LV-GCA patients tend to be younger, do not experience vision loss, are less likely to have cranial symptoms, and are more likely to have vascular findings [Schmidt WA et al. Rheumatology (Oxford) 2008]. The diagnosis of LV-GCA is challenging and typically takes 3 to 7 months from symptom onset. Diagnosis is based on careful clinical examination, with assessment of pulses and blood pressure in all four extremities, together with vascular imaging—either catheter-directed, magnetic resonance (MR), or computed tomography (CT) angiography [Grayson PC et al. J Rheumatol 2012].
Patients with LV-GCA tend to have more refractory disease than those with cranial symptoms, with more relapses and a greater reliance on steroids [Muratore F et al. Arthritis Rheum 2012 (abstr 2358)]. In upper extremity disease, ischemic complications are rare and most patients improve slowly without needing revascularization [Muratore F et al. Arthritis Rheum 2012 (abstr 2358); Schmidt WA et al. Rheumatology (Oxford) 2008]. By contrast, about one quarter of patients with lower extremity involvement develop critical leg ischemia [Czihal M et al. J Rheumatol 2012].
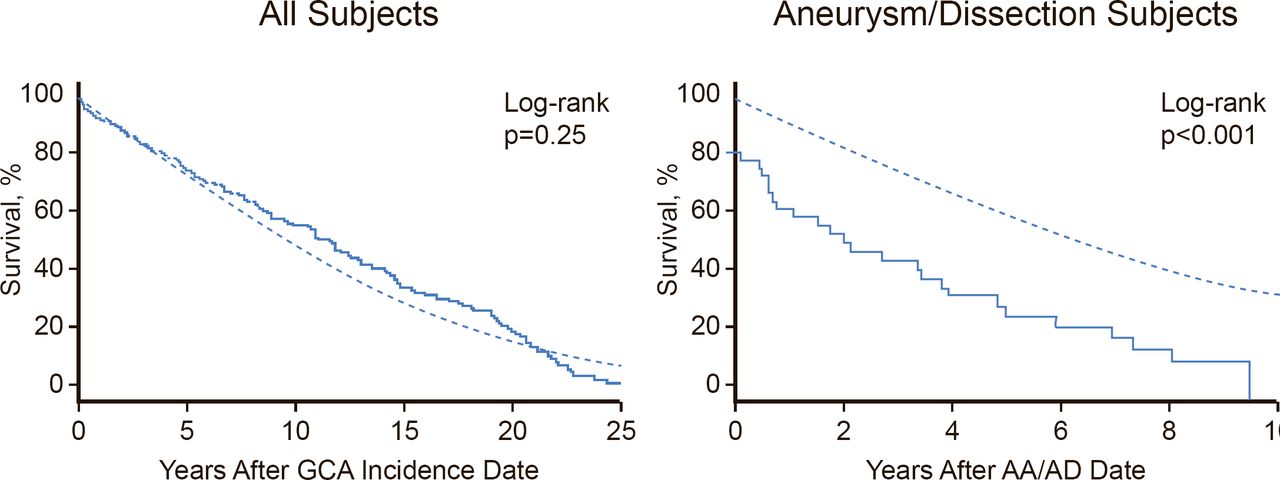
The aorta is involved in more than half of patients with GCA [Blockman D. Rheumatology (Oxford) 2008]. This is clinically significant because aortitis at the onset of GCA may predict subsequent aortic dilatation. Patients with GCA face a significantly increased risk of aortic aneurysm and the risk increases progressively over time. It is therefore important to monitor patients for aortic disease, even if they are asymptomatic, because aortic dissection or aneurysm in GCA patients significantly increases mortality (Figure 2) [Kermani TA et al. Ann Rheum Dis 2013]. There are limited data to support screening; however, Prof. Warrington stated that CT or MR angiography at baseline and repeated every 3 years seems “reasonable.”
TAKAYASU ARTERITIS
Eamonn Molloy, MD, MS, FRCPI, St. Vincent's University Hospital, Dublin, Ireland, reviewed the management of patients with TAK. This is a primary idiopathic LV vasculitis that typically affects patients under the age of 50 and is more common in women than in men [Kerr GS et al. Ann Intern Med 1994].

Survival of Patients With GCA or Aortic Aneurysm/Dissection Who Develop Large-Artery Stenosis
Reproduced from Kermani TA et al. Large-vessel involvement in giant cell arteritis: a population-based cohort study of the incidence-trends and prognosis. Ann Rheum Dis 2013;72(12):1989–94.
With permission from BMJ Publishing Group.
Assessment of disease activity is challenging in patients with TAK. Patients with TAK may be asymptomatic; symptoms, when present, may be systemic or related to arterial inflammation or arterial compromise [Maksimowicz-McKinnon K et al. Arthritis Rheum 2007]. There is no single diagnostic test; instead, diagnostic assessment requires consideration of the history, physical examination, laboratory tests, and imaging. Particularly early in the course of disease, physical exam may be normal and there is no established biomarker for TAK. New clinical features that develop during the course of follow-up may also relate to vascular damage, secondary atherosclerosis, medication side effects, intercurrent infection or other comorbidities rather than active TAK. Catheter-directed angiography was the historic gold standard for visualizing vascular lesions; it has now largley been supplanted by noninvasive modalities such as MR or CT angiography and positron emission tomography (PET) scanning [Grayson PC et al. J Rheumatol 2012].
In patients with established TAK, vascular imaging should be performed at least annually, even in the absence of symptoms. The detection of a new vascular lesion should prompt a thorough evaluation of the aorta and first-order branches as well as other vessels based on the patient's symptoms and history. Hypertension is a frequent complication of TAK and can go undetected because of peripheral arterial stenosis and present with end-organ damage. In some cases, catheter-directed angiography may be required to measure central aortic pressure.
Steroids are the mainstay of treatment of TAK, with prednisolone being started at 40 to 60 mg/day and gradually tapered. This brings about remission in ∼60% of patients, although relapses tend to occur during taper [Keser G et al. Rheumatology (Oxford) 2013]. Patients often need other drugs to counter the adverse effects of prednisolone [Maksimowicz-McKinnon K et al. Arthritis Rheum 2007]. Furthermore, most patients require other immunosuppressants to achieve disease control. Several drugs have shown efficacy in this setting, including methotrexate, azathioprine, and mycophenolate mofetil, but none have been evaluated in an RCT. Cyclophosphamide is reserved for use in immediately life-threatening disease [Maksimowicz-McKinnon K et al. Arthritis Rheum 2007].
The use of biologic therapies in TAK has been reported including anti-tumor necrosis factor drugs, tocilizumab and rituximab, with the greatest experience to date being with infliximab. However, to date, none of these has been evaluated in a RCT. The RCT Abatacept for Treating Adults with Giant Cell Arteritis and Takayasu's Arteritis [NCT00556439] evaluating the efficacy and safety of abatacept in TAK is currently recruiting; results will not likely be available before 2015.
SUMMARY
The information presented at the session reinforced the notion that managing LV vasculitis requires a highly skilled specialist and that much of current practice is unsupported by robust data. Furthermore, steroids are the mainstay of therapy and are hampered by poor tolerability, highlighting the urgent need to develop new treatment options.
- © 2013 MD Conference Express®
Tools









{kind=link}
{kind=link}
Table of contents
Cited By...
- No citing articles found.