

Summary
The Assessment of Pirfenidone to Confirm Efficacy and Safety in Idiopathic Pulmonary Fibrosis trial [ASCEND; King TE et al. N Engl J Med 2014] was a Phase 3, multinational, double-blind, placebo-controlled trial of pirfenidone in patients with idiopathic pulmonary fibrosis. This trial discusses findings.
- Pulmonary Clinical Trials
- Lower Respiratory Infections
- Pulmonary Clinical Trials
- Pulmonary & Critical Care
- Lower Respiratory Infections
The Assessment of Pirfenidone to Confirm Efficacy and Safety in Idiopathic Pulmonary Fibrosis trial [ASCEND; King TE et al. N Engl J Med 2014] was a Phase 3, multinational, double-blind, placebo-controlled trial of pirfenidone in patients with idiopathic pulmonary fibrosis (IPF). The findings were presented by Talmadge E. King, Jr, MD, University of California, San Francisco, San Francisco, California, USA.
IPF is a chronic, progressive, and irreversible lung disease characterized by pulmonary scaring that ultimately leads to pulmonary dysfunction and death. The prognosis is poor, with a median survival time from diagnosis of 2 to 5 years. Two of 3 prior Phase 3 studies with pirfenidone demonstrated a reduction in lung deterioration and progression-free survival (PFS) [Taniguchi H et al. Eur Respir J 2010; Noble PW et al. Lancet 2011]. On the basis of the inconsistent Phase 3 data, it was recommended by US regulatory authorities that the ASCEND trial be undertaken.
In the ASCEND trial, 555 eligible patients with IPF (summarized in Table 1) were randomized 1:1 to receive oral pirfenidone (2403 mg/day; n=278) or placebo (n=277) for 52 weeks. Clinical follow-up included assessments at Day 1 and Weeks 13, 26, 39, and 52 on therapy as well as 28 days after the last dose of study drug. The primary end point was the change in forced vital capacity (FVC; ≥10% predicted) or death at Week 52. Secondary end points were change in 6-minute walk distance (6MWD) at Week 52, PFS (time to first occurrence of death, confirmed ≥10% decline in FVC, or confirmed ≥50-m decline in 6MWD), dyspnea, all-cause mortality, and treatment-related mortality.
Eligibility Criteria
Baseline characteristics were well balanced between treatment groups, with a median age of 68 years, median FVC of 68% predicted, and 6MWDs ranging from 415 to 421 meters. A total of 461 patients (94%) completed the study (pirfenidone, n=261 [93.9%]; placebo, n=261 [94.2%]), and 55 patients (19.8%) in the pirfenidone group and 39 (14.1%) in the placebo group prematurely discontinued study treatment.
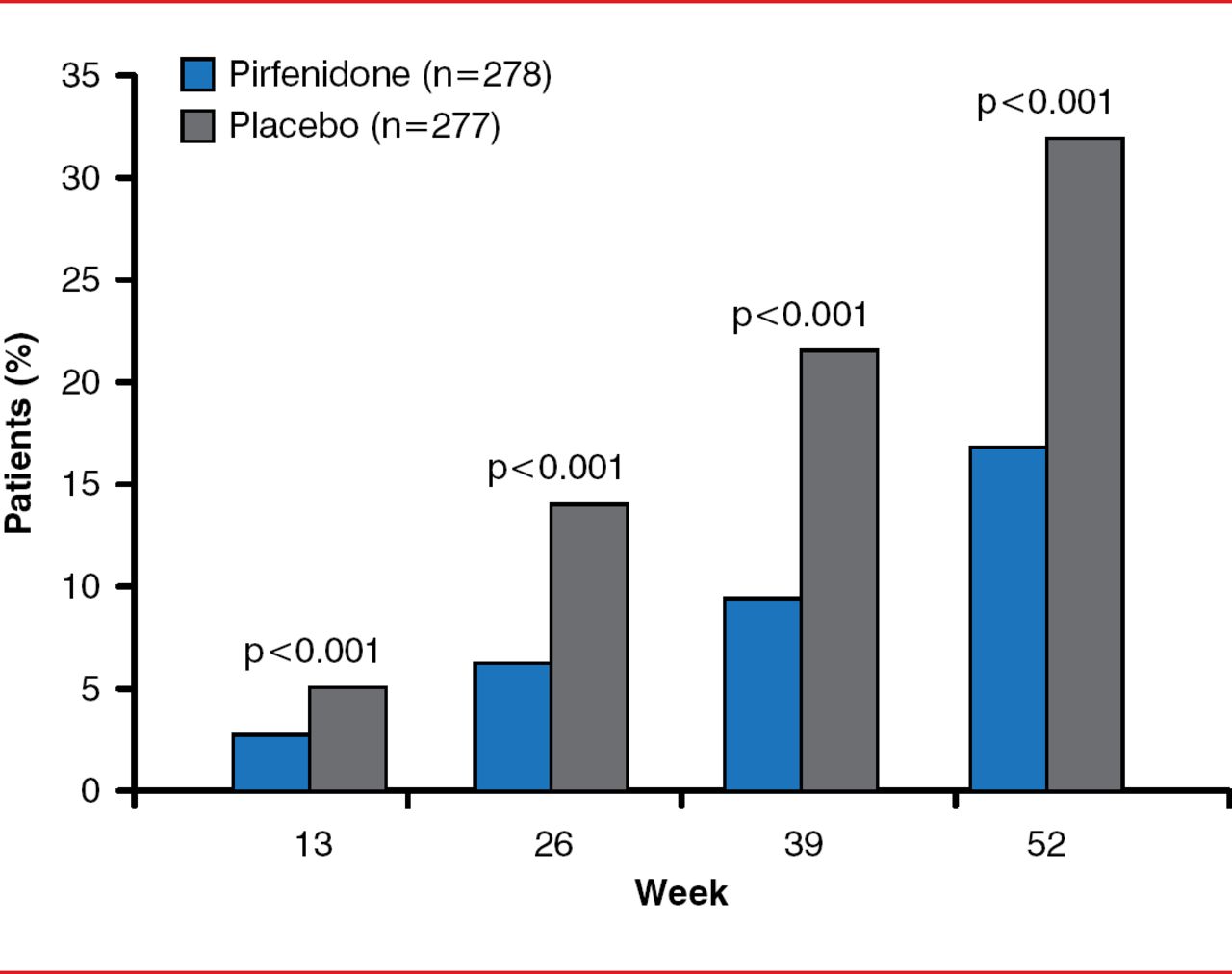
By Week 52, the primary end point, decline in FVC or death, was significantly reduced in the pirfenidone group by 47.9% compared with the placebo group (46 patients [16.5%] vs 88 patients [31.8%], p<0.001; Figure 1). By Week 52, there was a slower decline in FVC (relative difference of 45.1%, p<0.001), fewer subjects with decreased 6MWDs or death (p=0.04), and improved progression-free survival (HR, 0.57; 95% CI, 0.43–0.77; p=0.0001). Dyspnea scores were not significantly improved. Pirfenidone was associated with fewer deaths from any cause (11 [4.0%] vs 20 [7.2%], p=0.01) and death from IPF (3 [1.1%] vs 7 [2.5%], p=0.23); however, the study was not powered for this end point. A prespecified analysis with pooled data from the ASCEND and 2 Phase 3 CAPACITY trials found a 48% reduction in the risk for all-cause mortality in the pirfenidone group compared with the placebo group (HR, 0.52; log rank p=0.0107).

Primary End point of Decreased FVC or Death
Reproduced with permission from TE King, Jr, MD.
Pirfenidone was associated with fewer deaths from any cause (22 [3.5%] vs 42 [6.7%], p=0.01) and death from IPF (7 [1.1%] vs 22 [3.5%], p=0.006) [Nobel PW et al. Lancet 2011].
Treatment with pirfenidone showed a favorable safety profile and was generally well tolerated (Table 2).
Adverse Eventsa
The most common adverse events, with higher incidence in the pirfenidone group, were gastrointestinal (nausea, 36.0% vs 13.4%; dyspepsia, 14.7% vs 6.1%) and skin related (eg, rash; 28.1% vs 8.7%). These were typically mild to moderate in severity and could be reversed with treatment. Adverse events that prompted discontinuation occurred in 14.4% in the pirfenidone group and 10.8% in the placebo group, most commonly due to worsening of IPF (1.1% and 5.4% in the pirfenidone and placebo groups, respectively). Serious adverse events occurred in 19.8% and 24.9% of patients in the pirfenidone and placebo groups, respectively, and again most often involved worsened IPF (2.5% and 9.7%, respectively).
The authors concluded that pirfenidone was well tolerated and led to a reduction in disease progression or death for patients with IPF.
- © 2014 MD Conference Express®
Tools









{kind=link}
Table of contents
Cited By...
- No citing articles found.