

Summary
Despite advancements in medical device technology over the past several decades, safety issues remain a problem in diabetes technology, including glucose meters and insulin pumps. This article explores diabetes technology with specific topics including past and present devices, the evaluation of diabetes technology, and the US perspective on the evaluation of diabetes devices.
- Diabetes Mellitus
- Insulin
- Exclusive Article - For home page
- Diabetes Mellitus
- Endocrinology
- Diabetes & Metabolic Syndrome
- Insulin
- Exclusive Article - For home page
Despite advancements in medical device technology over the past several decades, safety issues remain a problem in diabetes technology, including glucose meters and insulin pumps. John C. Pickup, BM, PhD, King's College London School of Medicine, London, United Kingdom, introduced a session exploring diabetes technology with a discussion of past and present devices.
Early continuous subcutaneous insulin infusion (CSII) safety data from the 1980s suggested pump use resulted in less hypoglycemia and less increase in diabetic ketoacidosis compared with injection therapy. A 1980s survey showed that 25% of CSII pumps malfunctioned; 29% from drive failure and 14% from battery issues. In addition, 81% of patients reported infusion setting problems, of which 53% were due to blockage.
Modern insulin pump therapy is more complex and sophisticated. Some features include an alarm for malfunctions, software to aid assessment, and wireless connectivity to adjust settings. Although this complexity translates into greater opportunities for adverse events (AEs), clinical experience with pumps has greatly increased. In terms of safety, some premarket regulations have been developed, yet postmarket surveillance for safety and efficacy of insulin pumps is limited and is often not available to the public.
Despite the more advanced technology in modern insulin pumps, pump failures and AEs still occur frequently. In a survey of 640 new pumps from 4 different insulin pump manufacturers, 36% of pumps were reported to have any defect, 16% reported a complete pump failure, and 6.5% reported mechanical defects that required replacement [Guilhem I et al. Diabetologia 2009]. In a 10-year retrospective study of AEs reported to the United States Food and Drug Administration (FDA), 1594 AEs related to insulin pumps were reported between 1996 and 2005 and 13 deaths were documented [Cope JU et al. Pediatrics 2008]. However, this study lacked a matched control group and FDA verification of the accuracy and completeness of the reported AEs.
In a study of nonmetabolic complications of CSII, 96 patients with type 1 diabetes mellitus (T1DM) using a CSII for ≥6 months completed a questionnaire about demographics, infusion set type and duration, pump model, and frequency of complications. The rates of kinking, blockage, leaking, and malfunction are shown in Table 1.
Patient Survey: Insulin Pump Complications
Infusion-site complications such as lipohypertrophy (26.1%), infection (17.4%), bleeding or bruising (14.1%), pain or soreness (5.4%), and irritation or itchiness (5.4%) were reported. Interestingly, a common cause of CSII AEs is ultimately due to patient error, poor patient training, or patient compliance, such as accidental damage to the pump, poor infusion set connection, or missed bolus.
Lutz Heinemann, PhD, Science & Consulting, Düsseldorf, Germany, provided the European perspective on evaluating diabetes technology. A CE mark, required for market approval of a medical device in the European Union (EU), is provided by a Notified Body that is accredited to assess safety and performance, but not clinical effectiveness. However, a new framework for EU regulatory approval has been proposed, and a vote of the European Parliament is anticipated end of October 2013. One proposed change is basing approval on the associated risk of a medical device, such as insulin pumps, and in vitro diagnostics, such as blood glucose meters.
Some limitations of the current system, according to Prof. Heinemann, include the quality of the postmarketing surveillance system required of the manufacturer after obtaining a CE mark, the time required to identify an issue, and the type of event that prompts a physician to report it. Another limitation is that devices are not being withdrawn from the market even though they do not meet regulatory requirements—about 20% to 50% of blood glucose meters fall into this category.
Quality management systems by manufacturers are required and regulated by government authorities. The public cannot access reports or reviews of medical devices in the EU. Regulatory agencies perform regular manufacturing inspections; however, smaller companies are less likely to be inspected because of their size.
Safety issues associated with insulin pumps are the accuracy of the insulin infusion, bolus calculators (duration of insulin action), infusion catheters (for patients, not usually for manufacturers), and timing of occlusion alarms. Safety issues associated with insulin pens are the lack of independent head-to-head comparisons (most studies are a single manufacturer study of their device), and whether the accuracy is reduced in daily life.
The European Associate for the Study of Diabetes (EASD) is also addressing the issue of market approval and surveillance of diabetes devices. Some steps include holding an EASD diabetes technology meeting, a diabetes technology study group, supporting a registry of insulin pumps in Sweden, and the development of a statement proposing a new methodology for evaluating insulin pumps.
David B. Sacks, MB, ChB, National Institutes of Health, Bethesda, Maryland, USA, provided the US perspective on the evaluation of diabetes devices. The FDA classifies medical devices based on their associated risk. Class I (low risk) devices are exempt from formal review, and most Class II (moderate risk) devices, which include insulin pumps, are cleared by the FDA for clinical use. Class III (high risk) devices must undergo clinical trials by the manufacturer to establish safety and efficacy.
The steps to obtain FDA approval of a Class II medical device are the submission of a premarket notification, also called a 510(k), that establishes the new device is at least as safe and effective as a similar device in clinical use, followed by FDA evaluation of the intended use, performance, and labeling of the device. The FDA can require postmarketing surveillance by the manufacturer if the device is likely to cause serious AEs if it fails, will be used in a pediatric population, will be implanted >1 year, or is intended as a life-sustaining or life-supporting device. Reports of AEs are reviewed by the FDA, and are followed-up if they are serious or a pattern emerges.
All postmarketing surveillance reports in the United States are available to the general public. The Manufacturer and user Facility Device Experience, one such database, is a searchable list of manufacturers, device brand names, and date of reported events. Alerts from the reporting system or by a manufacturer may lead to product recalls.
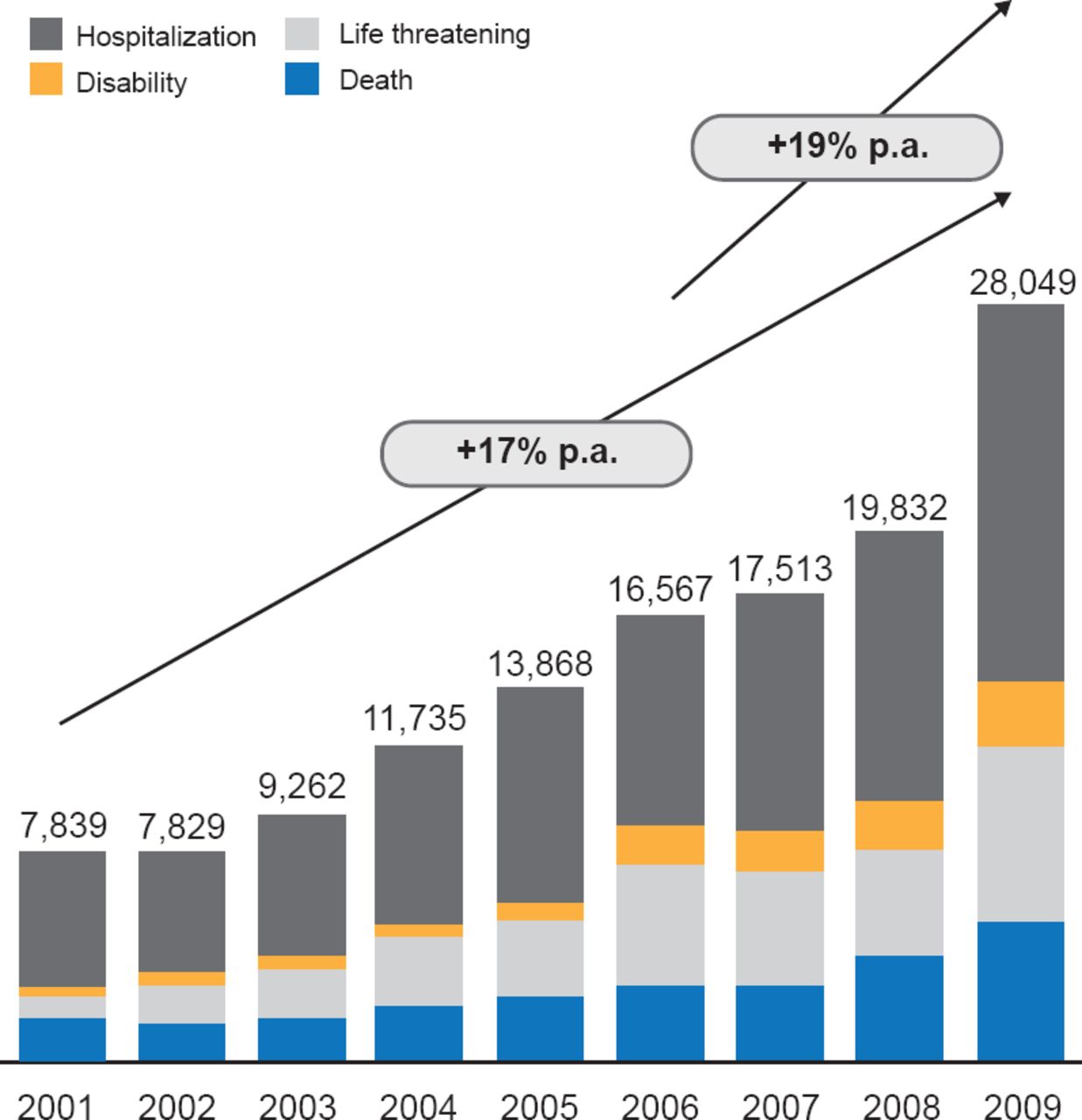
Notably, the number of serious and fatal medical device AEs rose about 17% per year between 2001 and 2009 (Figure 1). The greatest proportion of the serious AEs reported between 2005 and 2009 occurred with insulin pumps, and blood glucose test systems were the third most common cause.

Serious AE Associated With Medical Devices
Reproduced with permission from DB Sacks, MB, CHB.
Medical devices, particularly those used routinely in diabetes, are more complex and sophisticated, but AEs and device recalls remain and must be addressed.
- © 2013 MD Conference Express®
Tools









{kind=link}
Table of contents
Cited By...
- No citing articles found.