

Summary
A growing consensus links the loss of beta-cell mass with the development of type 2 diabetes mellitus. This article demonstrates that endoplasmic reticulum stress, induced by high levels of saturated free fatty acids, triggers a response that leads to eventual beta-cell apoptosis. This observation suggests one or more new targets for the therapeutic maintenance of beta-cell populations.
- endocrinology
- prevention & screening
- diabetes mellitus
A growing consensus links the loss of beta-cell mass with the development of type 2 diabetes mellitus (T2DM). A lecture that was presented by D.L. Eizirik, MD, PhD, Laboratory of Experimental Medicine, Université Libre de Bruxelles, Brussels, Belgium, demonstrates that endoplasmic reticulum (ER) stress, induced by high levels of saturated free fatty acids (FFAs), triggers a response that leads to eventual beta-cell apoptosis. This observation suggests one or more new targets for the therapeutic maintenance of beta-cell populations.
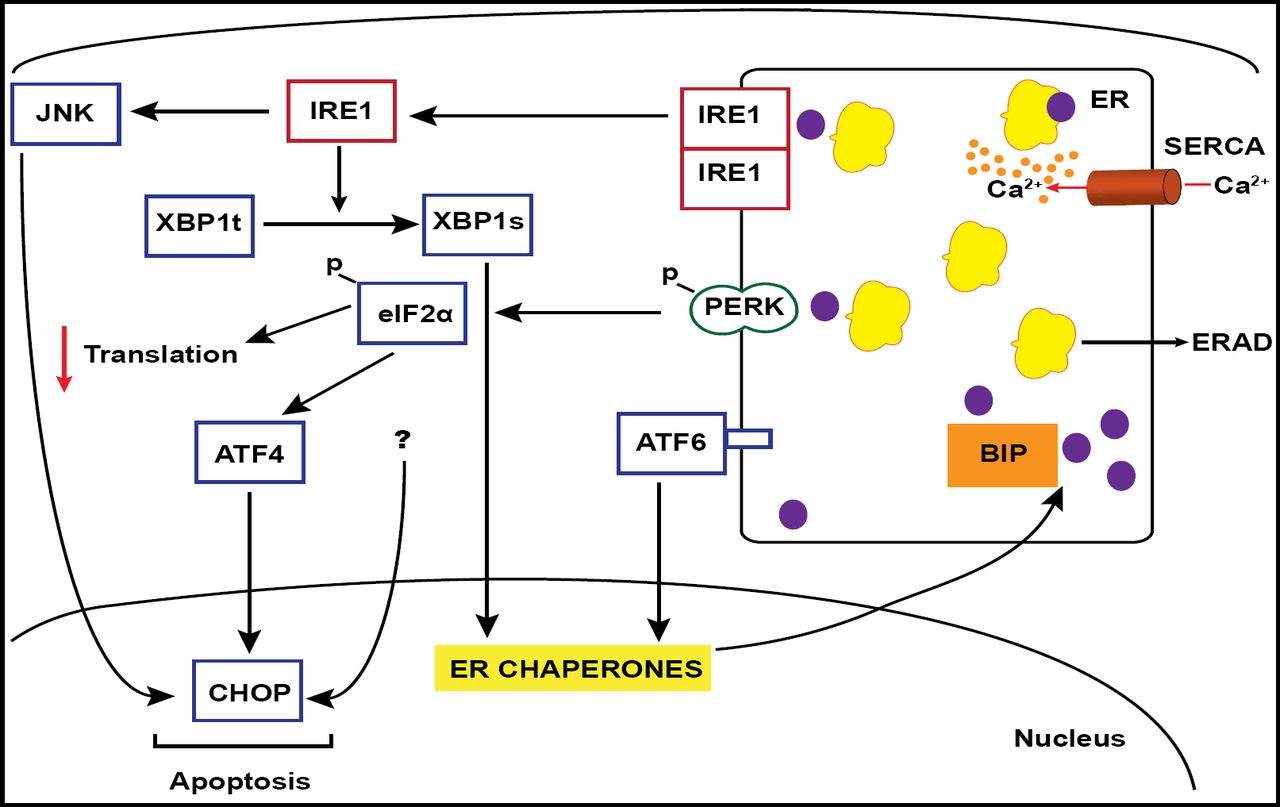
Recent evidence suggests that the ER stress response plays a role in the pathogenesis of diabetes through the induction of beta-cell apoptotic pathways. This stress occurs in the presence of unfolded or misfolded proteins in the lumen of the ER and triggers the unfolded protein response (UPR); the end result is an upregulation of molecular chaperones to aid in protein folding or, failing that, in the face of chronic stress, the initiation of programmed cell death, or apoptosis (Eizirik et al. Endocrine Reviews 2008).
Saturated free fatty acids are candidate inducers of ER stress in T2DM. An investigation by Cunha et al. in Eizirik's laboratory (J Cell Sci 2008) exposed INS-1E (insulinoma) cells, FACS-purified rat primary beta-cells, and human islets to the unsaturated fat oleate and the saturated fat palmitate. Results showed a differential ER response based on fat saturation.
Palmitate induced the signaling of the pathways of 3 ER membrane-bound proteins, IRE1, and ATF6 – which probably are pro-cell survival in this case – but signals through the membrane-bound protein PERK as well, which can trigger apoptosis through the proapoptotic elF2alpha/ATF4/CHOP signaling cascade (Figure 1). This observation was verified through the use of the reagent salubrinal, a phosphatase inhibitor that is known to render the gatekeeper elF2alpha constitutively active. In the presence of oleate, salubrinal elicited the same UPR expression profile and cell death in INS-1E cells and FACS-purified rat primary beta-cells as those seen with exposure to the saturated fatty acid palmitate (Cnop et al. Journal of Biological Chem 2007). These results were replicated in human islet cell preparations (Ladriere, Eizirik, and Cnop. Unpublished data).

ER Stress Signaling Cascade.
Palmitate also induces IRE1 signaling, promoting cell survival through downstream chaperone activation or, alternately, tipping the scales toward apoptosis through activation of Jun kinase (JNK). This association was shown using JNK inhibitors. In vitro work also was able to verify the palmitate/UPR/CHOP association by knocking down palmitate-induced expression with the use of CHOP siRNA and thus partially protecting beta-cells against palmitate-induced apoptosis (Cunha et al. J Cell Sci 2008).
Conversely, IRE1 signaling that promotes cell survival through the production of chaperones (eg, BiP) was validated through the use of BiP overexpression to rescue an insulin-secreting cell line from lipid-induced apoptosis (Laybutt et al. Diabetologia 2007).
A final association is to be made between ER stress and the dysfunction and loss of beta-cells, as seen in diabetes. Studies have shown that in the islets of db/db (diabetic) mice, numerous ER stress factors were unregulated – this also is true of human islet preparations (Laybutt et al. Diabetologia 2007). More to the point, both the proapoptotic CHOP expression and the ER area were increased in islets from patients with T2DM (Huang et al. Diabetes 2007; Marchetti et al Diabetologia 2007). Interestingly, islets from obese patients also show some increase in cytosolic CHOP expression. This, and additional evidence, suggests that ER stress may be a common mediator of both beta-cell death and insulin resistance in T2DM (Eizirik et al. Endocrine Reviews 2008).
- © 2008 MD Conference Express
Tools









{kind=link}
Table of contents
Cited By...
- No citing articles found.