

Summary
Studies have identified subclinical hypercortisolism in patients with adrenal incidentalomas, which is associated with greater rates of cardiovascular disease, type 2 diabetes mellitus, and mortality. This article discusses the genetics of bilateral macronodular adrenal hyperplasia, subclinical Cushing syndrome, as well as hypercortisolism in adrenal incidentaloma.
- adrenal disorders
Studies have identified subclinical hypercortisolism in patients with adrenal incidentalomas, which is associated with greater rates of cardiovascular disease, type 2 diabetes mellitus (T2DM), and mortality. Jerome Yves Bertherat, MD, PhD, Paris Descartes University, Paris, France, described the genetics of bilateral macronodular adrenal hyperplasia (BMAH), a cause of hypercortisolism. The development of BMAH has been associated with several known genes, including somatic mutations of GNAS [Weinstein LS et al. N Engl J Med 1991] and duplication of PRKACA (resulting in MAH) [Beuschlein F et al. N Engl J Med 2014], as well as genes associated with multiple tumor syndromes such as Menin, APC [Gaujoux S et al. Clin Cancer Res 2011], and fumarate hydratase [Matyakhina L et al. J Clin Endocrinol Metab 2005].
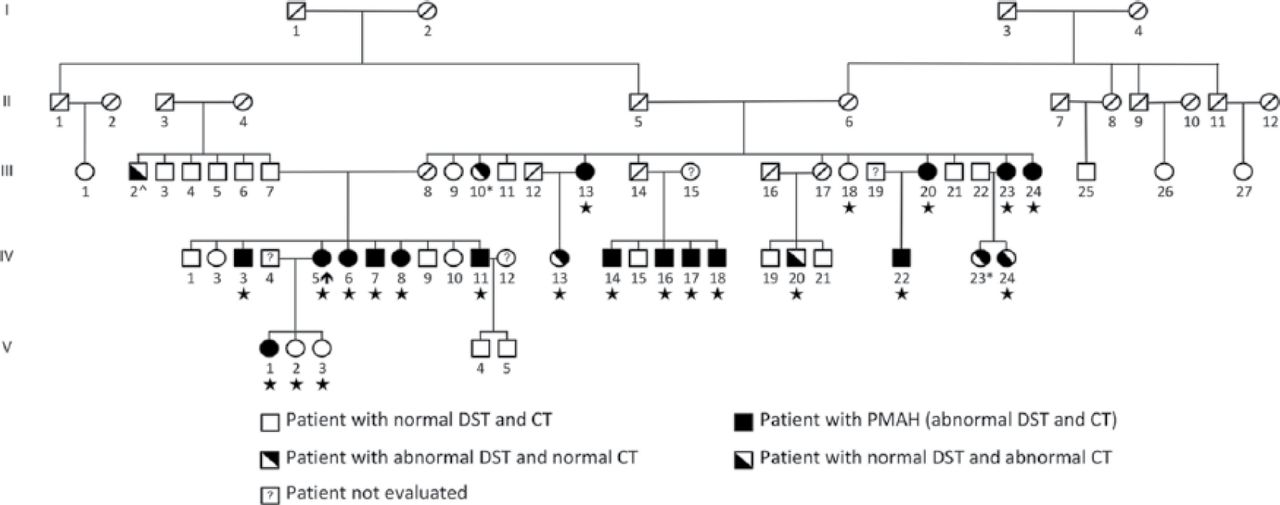
Prof. Bertherat hypothesized that 1 copy of an unknown gene receives a “hit,” or germline mutation, and later, a somatic mutation occurs in the second copy, resulting in the development of BMAH. To evaluate this hypothesis, the group searched for a gene that may be affected by a germline and somatic mutation. A single-nucleotide polymorphism array was used to perform a genome-wide screening for chromosomal aberrations by comparing leukocyte and BMAH tumor DNA. Loss of heterozygosity at chromosome 16p was detected in 24% of tumor samples from patients with BMAH [Assie G et al. N Engl J Med 2013]. Whole-genome sequencing of 5 leukocyte-tumor DNA pairs was undertaken and found that the armadillo repeat containing 5 (ARMC5) gene, located on chromosome 16p11.2, was mutated in 4 of 5 leukocyte samples and 3 of 5 tumor samples. Further analysis of the entire cohort of patients with BMAH demonstrated that 55% of patients harbored inactivating heterozygous ARMC5 mutations. In addition, recent studies have identified ARMC5 mutations in multiple families with BMAH, including a large family from Brazil (Figure 1) [Alencar GA et al. J Clin Endocrinol Metab 2014; Gagliardi L et al. J Clin Endocrinol Metab 2014].

Mutation of ARMC5 Resulting in BMAH in a Large Family
CT=computed tomography; DST=dexamethasone suppression test; PMAH=primary macronodular adrenal hyperplasia.
Reproduced from Alencar GA et al. ARMC5 mutations are a frequent cause of primary macronodular adrenal hyperplasia. J Clin Endocrinol Metab. 2014;99(8):E1501–E1509. With permission from The Endocrine Society.
A genotype-phenotype study in a cohort of European patients with BMAH who harbored wild-type or mutated ARMC5 found several meaningful, clinical differences. A significantly greater number of patients with mutated ARMC5 had clinical Cushing syndrome (71%) compared with patients with wild-type ARMC5 (35%; p = .006). In addition, patients with mutated ARMC5 had greater rates of hypertension (p = .009), T2DM (p = .093), and food response (p < .001). Serum adrenocorticotropic hormone (ACTH) levels were significantly higher in patients with mutated ARMC5 (p = .003), as were dexamethasone (1 mg) response (p < .001) and midnight plasma cortisol levels (p = .019). Furthermore, patients with mutated ARMC5 had greater body weight, larger adrenal size, and larger number of nodules identified by computed tomographic (CT) scan.
Prof. Bertherat concluded that ARMC5 is a novel tumor suppressor gene that is frequently mutated in patients with BMAH, likely causing impaired steroidogenesis; however, further studies are needed to assess the function of ARMC5 and its impact on patient management.
Massimo Terzolo, MD, S. Luigi Hospital, Orbassano, Italy, proposed that subclinical Cushing syndrome (SCS) in adrenal incidentaloma is a clinical entity. Adrenal incidentalomas are present in up to 5% of patients who undergo abdominal CT scans. Patients with adrenal incidentalomas do not typically display overt Cushing syndrome features but may have low-grade cortisol oversecretion. These patients may be considered to have SCS, in which specific symptoms of Cushing such as easy bruising, facial plethora, striae, and proximal myopathy are absent, but aspecific symptoms such as obesity, hypertension, T2DM, and osteoporosis are present [Nieman LK et al. J Clin Endocrinol Metab 2008].
A systematic review found that across many studies, the frequency of SCS ranged from 5% to 45% [Terzolo M et al. Eur J Endocrinol 2011]. Diagnostically, SCS may be identified by a failure to suppress on the dexamethasone suppression test (DST), elevated urinary free cortisol (UFC), low-suppressed ACTH, and elevated midnight serum cortisol (MSC). For example, 55% of patients with SCS have positive results on DSTs using a cutoff of 50 nmol/L, 8.8% have elevated MSC, 27% have suppressed ACTH, and 4.6% have an elevated UFC. However, the diagnosis can be challenging with the potential for false-positives, technical limitations of diagnostic testing, and the presence of a minimal degree of cortisol excess in these patients.
Recently, SCS has been associated with metabolic syndrome and osteoporosis; however, cause and effect has not yet been definitively proved because of small sample sizes, poor study design, and a lack of data from hard end points. One study in which SCS was defined as 1 mg-DST cortisol > 5 mg/dL found that patients with adrenal incidentaloma and SCS (5.5%) had greater rates of hypertension, T2DM, coronary heart disease, osteoporosis, and osteoporotic fractures compared with patients who did not have SCS (Table 1) [Di Dalmazi G et al. Eur J Endocrinol 2012].
Rates of Cardiovascular Disease Among Patients With Adrenal Incidentalomas
Another study found that patients with adrenal incidentalomas with 1 mg-DST cortisol > 1.8 mg/dL demonstrated a greater mean visceral/subcutaneous fat ratio and visceral/total volume fat ratio compared with patients who had lower cortisol levels [Debono M et al. J Clin Endocrinol Metab 2013]. A recent study with ≥ 5 years of follow-up found that 11.6% of patients had SCS, with an additional 8.2% of patients developing SCS during the follow-up period [Morelli V et al. J Clin Endocrinol Metab 2014]. The study defined SCS as 1 mg-DST cortisol > 5 mg/dL or at least 2 of the following: ACTH < 10 pg/mL, increased UFC, and 1 mg-DST cortisol > 3.0 mg/dL. In addition, at baseline, the presence of bilateral adenomas was greater in these patients, as was the presence of T2DM, hypertension, and cardiovascular events, rates of which increased substantially by the end of follow-up.
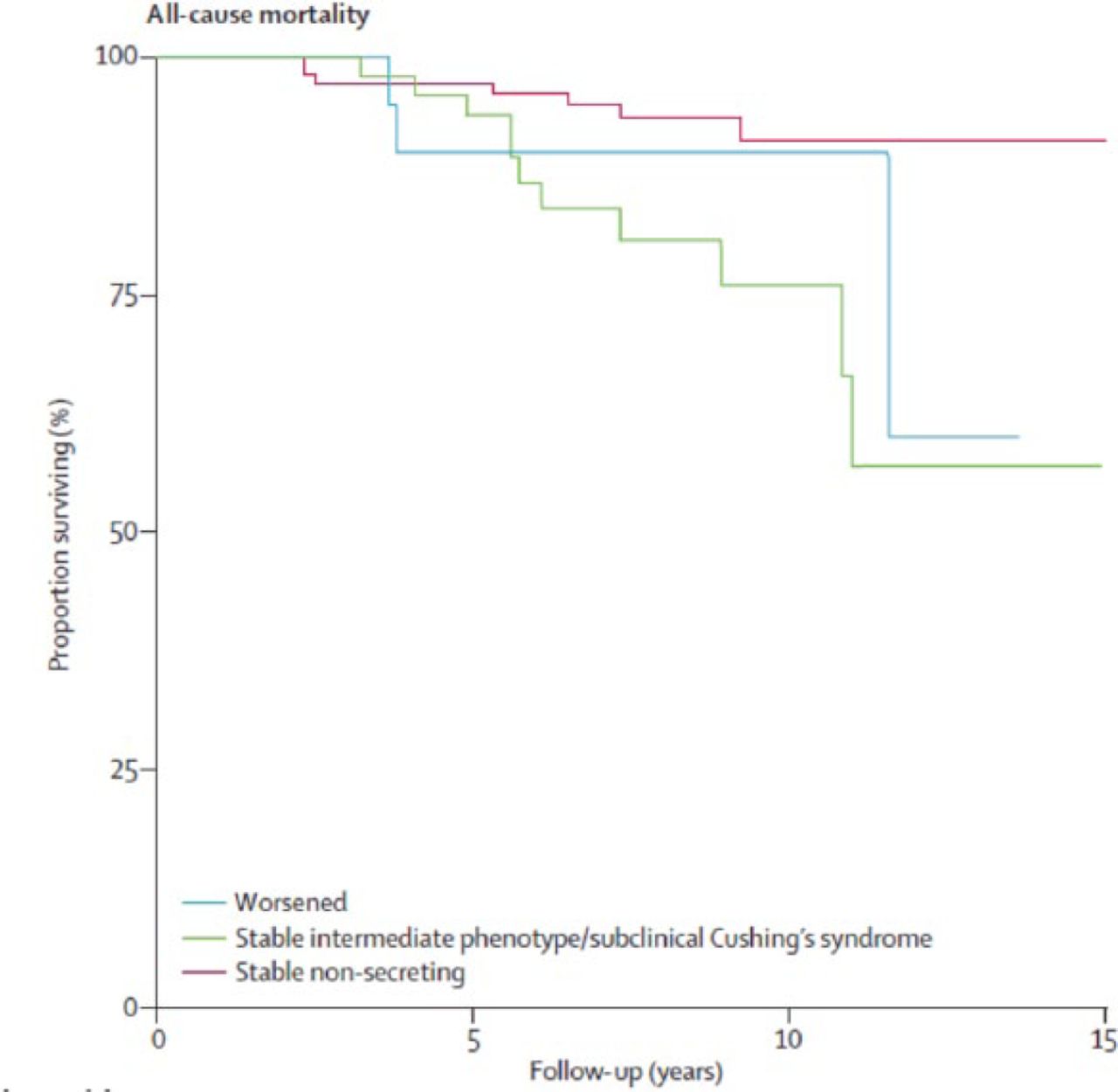
In a study of cardiovascular events and mortality in patients with adrenal incidentalomas, patients with SCS or an intermediate phenotype had greater rates of all-cause mortality (Figure 2) and cardiovascular mortality compared with patients with nonsecretory adenomas [Di Dalmazi G et al. Lancet Diabetes Endocrinol 2014].

All-Cause Mortality Among Patients With Adrenal Incidentalomas
Reproduced from Di Dalmazi G et al. Cardiovascular events and mortality in patients with adrenal incidentalomas that are either non-secreting or associated with intermediate phenotype or subclinical Cushing's syndrome: a 15-year retrospective study. Lancet Diabetes Endocrinol. 2014;2:396–405. With permission from Elsevier.
Prof. Terzolo concluded by stating that there is consistent evidence of autonomous cortisol secretion with evidence indicating an association between greater cortisol autonomy and clinical adverse effects; however, it remains difficult to determine which patients have adrenal autonomy that translates into clinically meaningful cortisol overproduction.
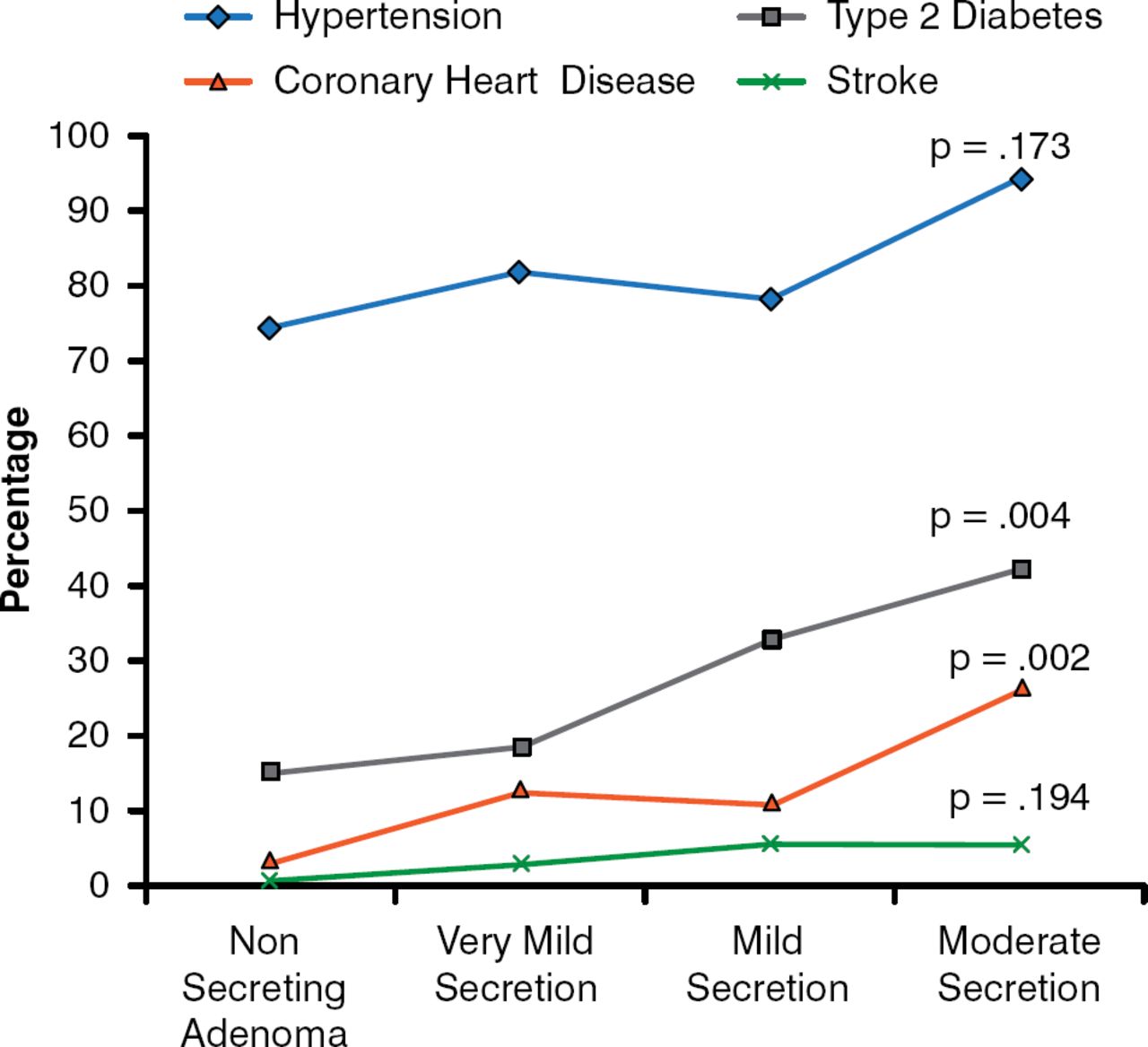
Miguel Debono, MD, MRCP, PhD, University of Sheffield, Sheffield, United Kingdom, discussed whether hypercortisolism in adrenal incidentaloma should be treated. The dilemma in treating these patients is that although SCS is associated with cardiovascular risk and events, osteoporosis, and mortality, many diagnostic tests lack specificity, and therefore false positives are possible. Evidence-based data indicate that even very mild to mild secreting phenotypes are associated with an increased risk for cardiovascular disease, including coronary heart disease, as well as T2DM (Figure 3) [Di Dalmazi et al. Eur J Endocrinol 2012]. Accumulated visceral fat also occurs more frequently in patients with mild hypercortisolism when compared with patients with nonsecretory adenomas [Debono M et al. J Clin Endocrinol Metab 2013].

Cardiovascular Risk Factors Among Patients With Adrenal Incidentalomas
Reproduced with permission from M. Debono, MD. Adapted from Di Dalmazi et al. Progressively increased patterns of subclinical cortisol hypersecretion in adrenal incidentalomas differently predict major metabolic and cardiovascular outcomes: a large cross-sectional study. Eur J Endocrinol. 2012;166:669–677.
Moreover, in both pre- and postmenopausal women with adrenal incidentalomas, mild hypercortisolism is associated with a greater number of fractures [Chiodini I et al. J Clin Endocrinol Metab 2004], and recent data show that hypercortisolism may be associated with increases in all-cause and cardiovascular mortality compared with patients with nonsecreting adenomas [Di Dalmazi G et al. Lancet Diabetes Endocrinol 2014].
However, surgical studies in patients with adrenal incidentalomas and hypercortisolism, diagnosed using conventional tests, show conflicting results, suggesting difficulties with diagnosis; in some cases, no improvement is observed after surgery, raising the question of false positives, and in other instances, although some patients are diagnosed with “nonfunctioning” adenomas, they do still improve after surgical intervention, indicating that tests may lack sensitivity for milder cases [Chiodini I et al. J Clin Endocrinol Metab 2010]. Therefore, Dr. Debono called for improved diagnostic strategies, which would require longitudinal, prospective, observational, and interventional studies to identify glucocorticoid biomarkers. In addition, randomized, prospective trials evaluating surgical intervention are also needed.
The presence of hypercortisolism in patients with adrenal incidentalomas is becoming more recognized, despite the absence of specific Cushing syndrome symptoms. However, the dilemma of surgical intervention in these patients continues, because of the risk associated with the disease and the potential for false positives.
- © 2014 MD Conference Express®
Tools









{kind=link}
{kind=link}
{kind=link}
Table of contents
Cited By...
- No citing articles found.