

Summary
Genetic breakthroughs have implications for both Parkinson disease and epilepsy. Evidence suggests that the monoclonal antibody Syn303 may represent a therapeutic advance in treating Parkinson disease. The discovery of genetic mutations and variants in families and others with epilepsy is providing groundbreaking insights regarding potential treatment options and surgical considerations.
- Parkinson disease
- epilepsy
- genetics
- monoclonal antibodies
- Syn303
- α-synuclein
- FFEVF
- DEPDC5
- mTOR
- neurology clinical trials
- extrapyramidal & movement disorders
- neurology genomics
At a symposium on the impact of new genetic insights on various neurologic disorders, 2 speakers highlighted the potential impact of these concepts on the diagnosis and treatment of Parkinson disease (PD) and epilepsy.
Virginia M.-Y. Lee, PhD, University of Pennsylvania School of Medicine, Philadelphia, PA, USA, discussed a possible role for monoclonal antibodies (mAbs) in the treatment of PD. The highly soluble protein α-synuclein is a major component of Lewy bodies, the protein-containing inclusions that are the pathologic hallmark of idiopathic PD [Volpicelli-Daley LA et al. Neuron. 2011], and it has been implicated in the pathogenesis of several neurodegenerative diseases, due most likely to an improper folding of the molecule [Saleh H et al. Transl Neurodegener. 2015].
The disease process is thought to occur via the uptake of improperly folding of the protein, which can then multiply by recruiting endogenously expressed counterparts. These then travel from affected cell to healthy cells and induce pathology throughout the nervous system. In the case of PD, the pathology starts in the medulla and olfactory bulb and then spreads through other parts of the brain and ultimately to the cerebral cortex [Braak H et al. Neurobiol Aging. 2003]. This type of stereotypical transmission suggests the involvement of a transmissible agent, such as α-synuclein.
Data suggest that synthetic seeding—the injection of α-synuclein preformed fibrils (PFFs) to initiate conversion of endogenous α-synuclein—begins the process of forming Lewy body inclusions in mice, rats, and nonhuman primates. This “seeded” pathology progressively expands, leading to α-synuclein inclusions that drive the selective loss of dopaminergic neurons in the substantia nigra pars compacta. This neuronal loss results in behavioral impairments suggestive of PD in humans [Luk KC et al. Science. 2012]. According to Dr Lee, this transmission of α-synuclein is a potential mechanism of PD pathogenesis.
Dr Lee then went on to discuss the possible role of immunotherapy in decelerating the neuronal losses caused by the α-synuclein inclusions. Based on the premise that blocking uptake of α-synuclein seeds with mAbs might halt cell-to-cell transmission of α-synuclein pathology, the mAb known as Syn303 was chosen for this proof-of-concept study [Tran HT et al. Cell Rep. 2014 ].
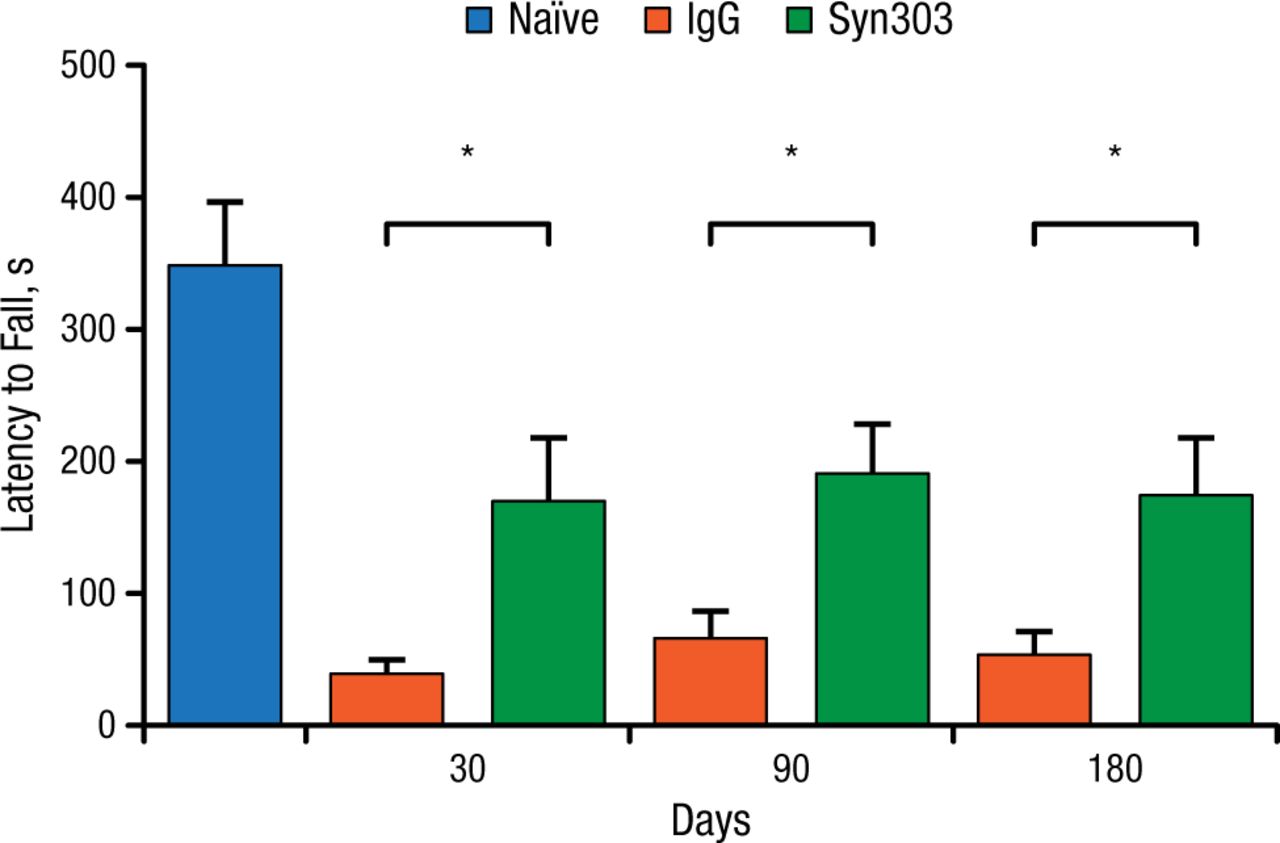
Dr Lee and her team used a sporadic PD mouse model to test the above hypothesis. They injected the dorsal striatum of the mice with α-synuclein PFFs, and the mice were then treated with either Syn303 or a control agent once a week. The outcome of interest was whether treatment with the mAb would be associated with reduced motor deficits based on results of a wire hang test at 30, 90, and 180 days. At all time points, the treated animals showed significant improvement on the wire hang test (P < .05; Figure 1).

Syn303 Wire Hang Performance, Wild-type Mice
IgG, immunoglobulin G.
*P < .05.
Adapted from Tran HT et al. α-Synuclein immunotherapy blocks uptake and templated propagation of misfolded α-synuclein and neurodegeneration. Cell Rep. 2014;7:2054-2065.
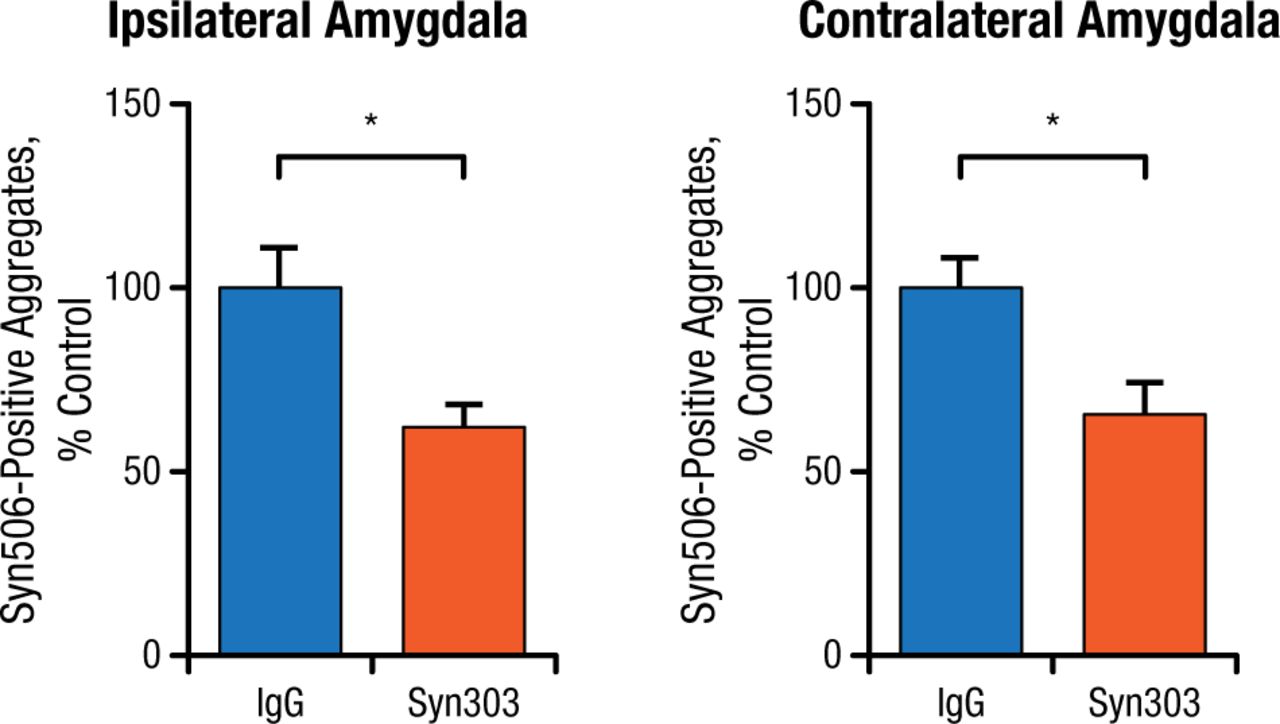
Prevention studies were done by injecting PFFs into normal mice as a control and then immediately adding Syn303. The control group showed PD pathology in multiple brain areas over time, while the mice treated with Syn303 showed significantly reduced pathology in the same areas (Figure 2).

Treatment With Syn303 Reduces Pathologic Spread of Syn506-Positive Aggregates
IgG, immunoglobulin G.
*P < .01.
Adapted from Tran HT et al. α-Synuclein immunotherapy blocks uptake and templated propagation of misfolded α-synuclein and neurodegeneration. Cell Rep. 2014;7:2054-2065.
In conclusion, Dr Lee emphasized that in a specific mouse model, mAbs appear to block uptake of misfolded endogenous α-synuclein and reduce the formation of α-synuclein inclusions that are the pathologic basis of PD in humans. This may represent a therapeutic advance in treating PD and Lewy body disease.
The second presentation focused on the genetic causes of familial focal epilepsies. Ingrid E. Scheffer, MBBS, PhD, University of Melbourne, Melbourne, Australia, began by highlighting an Australian family with a history of multiple members with familial focal epilepsy with variable foci (FFEVF), a focal epilepsy emanating in the frontal, temporal, or parietal region of the brain, with seizure onset ranging from infancy to adulthood. First described in 1998, FFEVF is unusual because the majority of epilepsies cause seizures that originate from only 1 region of the brain.
In the period between 1998 and 2012, 2 new children were born into this family, with both epilepsy and autism spectrum disorder. Exome sequencing subsequently discovered a mutation in the DEPDC5 gene. There are now 8 families in the literature with FFEVF; a recent study reports that 7 of the 8 carry the DEPDC5 mutation [Dibbens LM et al. Nat Genet 2013] but with a penetrance of only 66%. This same study also identified families with too few members to make a diagnosis of FFEVF. Among these smaller families, 12% also had DEPDC5 mutations. The breakdown of the regions where the seizures occurred is shown in Table 1.
Breakdown by Seizure Region in 95 People With DEPDC5 Mutations
Dr Scheffer went on to discuss the reason why different family members might have different focal epilepsies. Data suggest that DEPDC5 mutations interrupt the GATOR1 complex and activate the mammalian target of rapamycin (mTOR) pathway, which represses protein synthesis and autophagy [Bar-Peled L et al. Science. 2013]. Because the mutations found in patients with tuberous sclerosis also occur along this same pathway and can be treated with mTOR inhibitors such as everolimus and sirolimus, there is interest whether these drugs will also help patients with FFEVF. Additionally, it is possible that some patients with DEPDC5 mutations may have malformations of cortical development similar to the 2-hit hypothesis suggested in tuberous sclerosis. Dr Scheffer also suggested that a patient who has a genetic focal epilepsy with the DEPDC5 mutation may still benefit from surgical resection.
Dr Scheffer emphasized some of the work that is ongoing in finding genetic variants in children with photosensitivity [Galizia EC et al. Brain. 2015] and in children who have absence epilepsy with eyelid myoclonia. This work is likely to provide the basis for personalized medicine, such as the results from a 2014 study suggesting that quinidine might provide a benefit for treating patients with a particular type of severe epilepsy who had a KCNT1 mutation [Milligan CJ et al. Ann Neurol. 2014]. In closing, she reminded attendees that genetic counseling is a crucial component of care for patients and families who have a known genetic defect or variant.
- © 2015 SAGE Publications
Tools









{kind=link}
{kind=link}
Table of contents
Cited By...
- No citing articles found.