

Summary
Sickle cell disease can lead to stroke, which affects about 40% of those with the disease by mid-adolescence. The cognitive damage can lessen IQ and diminish quality of life. Research is leading to helpful strategies, and has also revealed details of the arterial accumulation of the affected red blood cells.
- silent stroke
- children
- adolescents
- biology
- physiology
Silent Strokes
Michael R. DeBaun, MD, MPH, Vanderbilt University Medical Center, Nashville, Tennessee, USA, discussed silent strokes in children and adolescents with sickle cell disease (SCD).
From 1990 to 1999, the incidence of SCD in the United States was 1 in every 2474 live births overall [Kanter J, Kruse-Jarres R. Blood Rev. 2013], which exceeds that of primary congenital hypothyroidism (1:3044) and cystic fibrosis (1:3000). The incidence for African Americans was markedly higher (1:400 live births). The implementation of newborn screening at the state level in 1988 was key; a study at that time by Vichinksy and colleagues showed that the mortality attributed to SCD decreased from 8% to 1.8% with newborn screening. Figure 1 illustrates the relative increase in survival [Quinn CT et al. Blood. 2010].

Improved Survival of Sickle Cell Disease
Adapted from Quinn CT et al. Improved survival of children and adolescents with sickle cell disease. Blood. 2010;115:3447-3452. With permission of American Society of Hematology.
The link between so-called silent strokes, which occur without any awareness in the person who is affected, has become recognized since only the 1990s. The cerebral damage inflicted by silent strokes can blunt academic performance and quality of life. The cerebral insult occurs early in life: by age 6 years, nearly one-third of those with SCD will have suffered at least 1 stroke, with deleterious effects apparent even before age 2 years.
The damage caused by a silent stroke includes diminished cognitive function, evident as decreased full-scale IQ [King AA et al. Am J Hematol. 2014]. Cognitive function may further diminish, given the progressive nature of silent strokes with time [Miller ST et al. J Pediatr. 2001]. Blood transfusions are valuable in reducing the risk of progression of silent cerebral infarcts [DeBaun MR et al. N Engl J Med. 2014; King AA et al. Pediatr Blood Cancer. 2008]. Socioenvironmental factors, including level of parental education and household income, can influence cognitive improvement [King AA et al. Am J Hematol. 2014]. With the life span of those with SCD now extended to adulthood, the consequences of the disease in the decades following childhood have become important to understand.
Dr DeBaun’s interests in SCD have included Nigeria, the country with the largest burden of the disease, with 150 000 newborn cases annually, compared to 1400 in the United States. Among Nigerian children and adolescents, the prevalence of strokes and silent strokes is estimated to be 15 000 and 45 000 per birth cohort year, respectively. The ongoing SPIN study [Galadanci NA et al. Pediatr Blood Cancer. 2014], funded by the US National Institute of Neurological Disorders and Stroke, is assessing the feasibility of low-dose hydroxyurea (the only approved drug for SCD) in children with elevated transcranial Doppler-determined blood velocity in lowering this velocity and so lessening the risk of stroke. This strategy could enable effective treatment in countries where blood transfusions are not economically feasible.
Biological and Physiologic Aspects of SCD
Robert Hebbel, MD, University of Minnesota, Minneapolis, Minnesota, USA, addressed biological and physiologic issues of SCD. The root of SCD is hemoglobin, the protein responsible for ferrying oxygen from the lungs throughout the body. SCD is caused by a single-point mutation on the β-globin subunit of hemoglobin, resulting in a mutant form of hemoglobin known as sickle hemoglobin (HbS). The sickle mutation confers an abnormal property on the HbS that causes it to polymerize when switched from the oxygenated to deoxygenated conformation, which causes the characteristic sickling of the red blood cells (RBCs). This is a reversible mutation; should the HbS become reoxygenated, the sickled RBC returns to its normal shape. As demonstrated by Dr Hebbel and colleagues in decades of research, the RBCs of a patient with SCD become sticky and accumulate in blood vessel of limbs and organs. The result is pain, organ damage, anemia, and increased risk of infection.
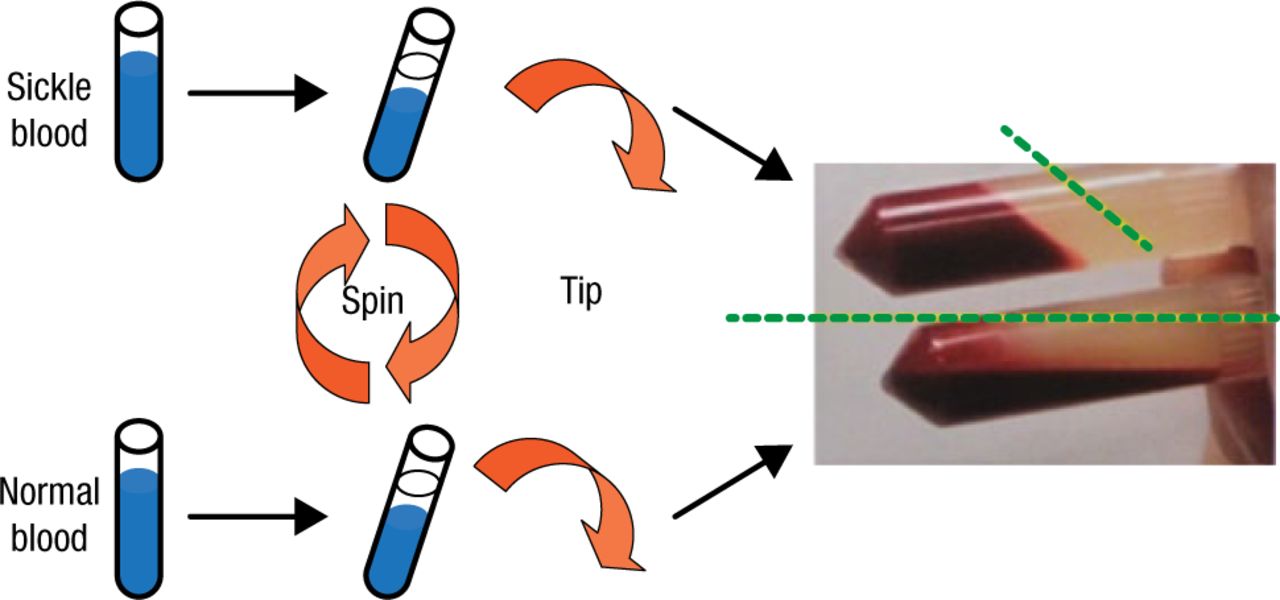
Research into the altered nature of RBCs in SCD that drives their vascular accumulation began with the serendipitous observation, first described in 1978 by Dr Hebbel and colleagues, of the tendency of the cells to aggregate and become less capable of flow when compared with normal RBCs (Figure 2).

Demonstration of the Altered Behavior of Red Blood Cells in Sickle Cell Disease
Reproduced with permission from R Hebbel, MD.
Subsequent in vitro studies by Dr Hebbel and others in the 1980s revealed the tendency of the abnormal red erythrocytes to form rosettes with endothelial cells and the correlation of the degree of cell adhesion with the severity of vascular occlusion. The association between sickle RBCs and endothelial cells is driven by an associative force that is stronger than that exhibited by normal RBCs, which produces more erythrocyte–endothelial cell contacts. The consequence is accumulation of the RBCs along the walls of the microvasculature immediately following the capillaries, which leads to vaso-occlusion.
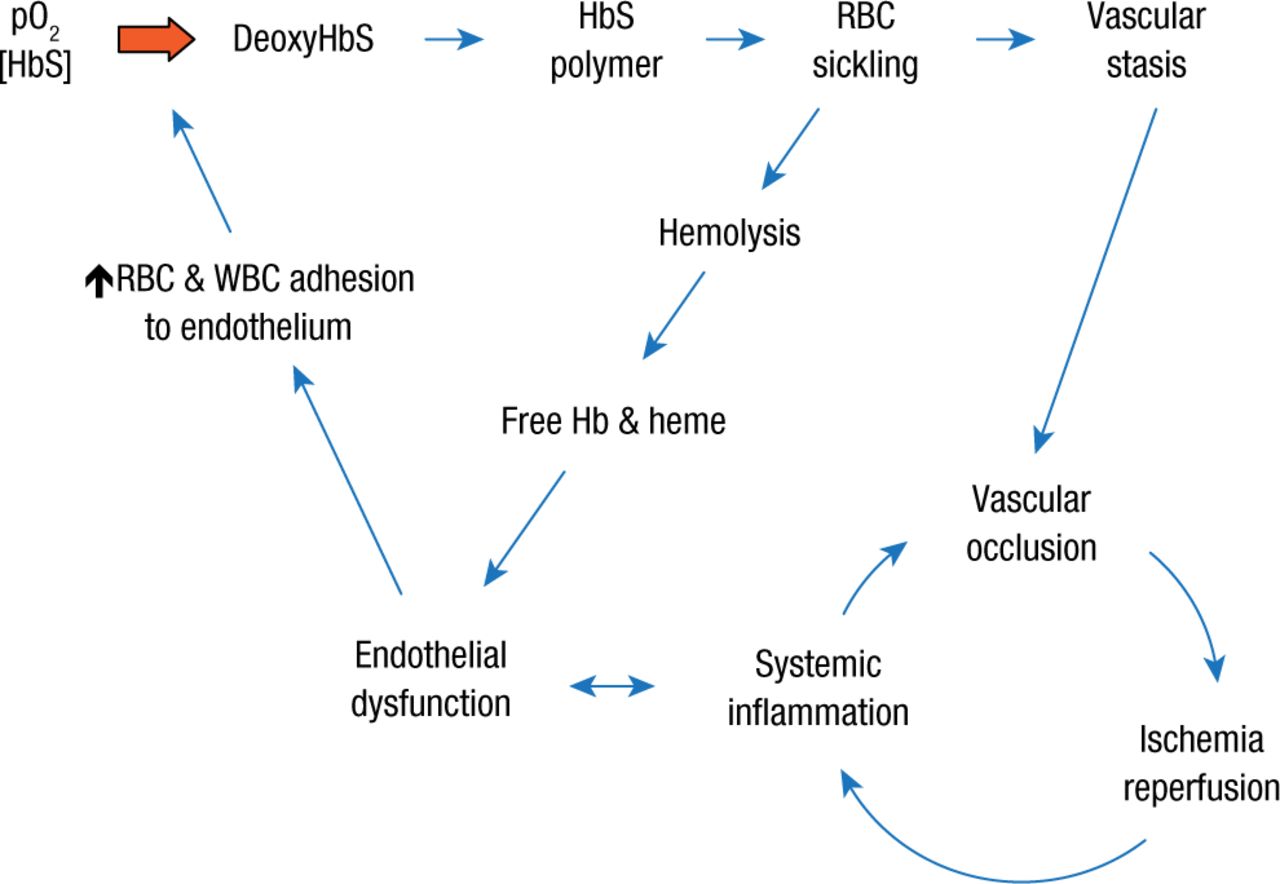
Normally, RBCs flow uniformly through the vessels; however, studies with a mouse strain that produces HbS have shown that the more pronounced surface attraction of the sickle erythrocytes produces more of a rolling motion along the vessel walls, which can lead to adherence and piling up of more erythrocytes. The result is a vicious cycle in which vascular occlusion drives ischemia reperfusion and systemic inflammation that in turn fuels occlusion and an overall vicious cycle in which systemic inflammation promotes endothelial dysfunction, which in turn encourages endothelial adhesion of both red and white blood cells (Figure 3).

The Vicious Cycles of Sickle Cell Disease
Hb, hemoglobin; HbS, hemoglobin S; pO2, partial pressure of oxygen; RBC, red blood cell; WBC, white blood cell.
Reproduced with permission from R Hebbel, MD.
Sickle cell adhesion is also influenced by platelets, increased expression of von Willebrand factor by activated endothelial cells, endothelial inflammation, and perhaps even endothelial damage caused by exposure to tobacco smoke.
Antiadhesive therapeutic studies have revealed that the inhibition of P-selectin on the endothelial cell surface improves blood flow, while inhibition of E-selectin (another adhesion molecule, which is expressed on the surface of cytokine-activated endothelial cells) does not. The findings support the view that increased expression of P-selectin in SCD mediates the adhesion of sickle erythrocytes to the endothelium. Other antiadhesive strategies are focusing on disrupting cell signals that mediate the association between sickle erythrocytes and adhesion molecules.
Whether the vascular accumulation of sickle erythrocytes involves capillaries is unclear. If this does occur—and whether it involves different mechanisms and so could require a different therapeutic approach—is also unknown.
- © 2014 SAGE Publications
Tools









{kind=link}
{kind=link}
{kind=link}
Table of contents
Cited By...
- No citing articles found.