

Summary
Genetic variants identified during the study of diabetes can be used as a tool to understand the cellular and molecular mechanisms that underlie the pathogenesis of diabetes. These variants can then be used to identify biomarkers, develop new therapies, and stratify patients to the most effective treatments, which is discussed in this article.
- Diabetes Mellitus
- Prevention & Screening
- Diabetes Mellitus
- Endocrinology
- Diabetes & Metabolic Syndrome
- Prevention & Screening
For the past 20 years, Anna L. Gloyn, DPhil, Oxford Centre for Diabetes Endocrinology and Metabolism, University of Oxford, Oxford, United Kingdom, has been studying genetic variants identified during the study of diabetes as a tool to understand the cellular and molecular mechanisms that underlie the pathogenesis of diabetes. Her goal is to identify biomarkers, develop new therapies, and stratify patients to the most effective treatments.
Evidence that this approach can be successful comes from a 2004 study that identified heterozygous activating mutations in the gene-encoding Kir6.2 subunit of the adenosine triphosphate (ATP)-sensitive potassium channel in pancreatic β cells as the cause of permanent neonatal diabetes [Gloyn AL et al. N Engl J Med. 2004]. Consequently, the β cells of these patients were unable to secrete insulin in response to glucose. This knowledge led to successful treatment of these patients with sulfonylureas, which target the KATP channel and restore insulin secretion.
Genome-wide association studies have been very successful in identifying regions of the genome robustly associated with type 2 diabetes mellitus (T2DM) risk. However, most of these signals map to regions of the genome that do not code for proteins, leaving uncertainty regarding the transcript and the proteins through which they exert their effect on diabetes. The current challenge, Prof Gloyn noted, is to move from the approximately 80 associated regions of the genome and identify the target proteins that will unlock the biology of diabetes.
Prof Gloyn then discussed the role coding variants, which are independently associated with diabetes risk or glycemic traits and independent from these genome-wide association study signals, could play in identifying these proteins. She discussed published data regarding a region of the genome associated with fasting plasma glucose (FPG) levels in people without diabetes. Two strong biological candidate genes close to the noncoding variant were identified as being possibly responsible [Bouatia-Naji N et al. Science. 2008; Chen WM et al. J Clin Invest. 2008]. The first candidate gene was G6PC2, which encodes for the islet-specific glucose-6-phosphatase catalytic subunit, known to catalyze the reverse reaction mediated by the pancreatic β-cell sensor glucokinase. The second was ABCB11, which encodes for an ATP-binding cassette transporter and for part of the bile salt export pump. ABCB11 was also considered a strong candidate because changes in bile salt secretion and bile acid pools can influence lipid and glucose metabolism.
The International GoT2D and T2D-GENES consortia performed exome sequencing in about 6500 patients with T2DM and a similar number of controls drawn from 5 different ancestries to obtain a more complete picture of the coding variation and to potentially resolve the uncertainty as to which of the 2 genes is responsible for the differences in FPG. This exome sequencing was supported by genotyping from more than 80 000 individuals of European ancestry, including 33 000 nondiabetic individuals for whom FPG levels were available. Two coding variants identified in G6PC2 were significantly associated with FPG levels, but no coding variants were identified for ABCB11. Coding variants in G6PC2 resulted in unstable proteins, suggesting that reduced G6PC2 protein in human pancreatic islets leads to reductions in FPG.
Prof Gloyn then discussed whether the T2DM-associated variants could alter the amount of gene transcript produced and thus whether the causal genes could be identified by this route. The majority of T2DM risk variants identified so far work through islet dysfunction [Voight BF et al. Nat Genet. 2010]. Using RNA and DNA extracted from human islets of 140 donors, Prof Gloyn and her colleagues determined that 1 of the T2DM risk variants influences ZMIZ1 expression. Although the complete role of ZMIZ1 in the pancreatic islet is still unclear, we now know that ZMIZ1 is localized specifically to the islets in the human pancreas and is expressed at higher levels in islets from T2DM donors than from those without T2DM.
While modulation of target proteins could potentially decrease T2DM risk, it could also have negative effects. Prof Gloyn and her colleagues have been pursuing the question of whether human genetics can identify these effects.
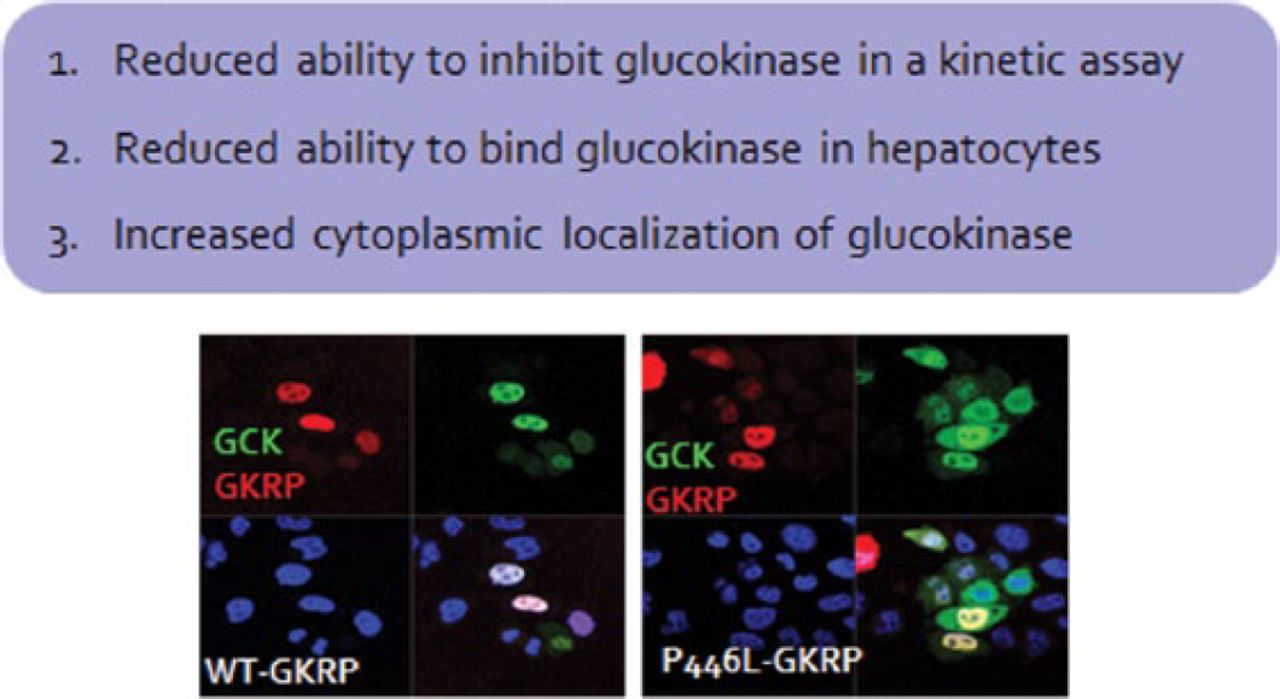
A 2008 study identified a coding variant (p.P446L) that resides in the glucokinase regulatory protein (GKRP) gene that is responsible for altered glucose levels, T2DM risk, and triglyceride levels [Orho-Melander M et al. Diabetes. 2008]. The allele that reduces glucose levels and T2DM risk also increases triglyceride levels. This is likely due to the reduced ability of the GKRP-P446L protein to bind glucokinase in hepatocytes, leading to increased amounts of glucokinase in the cytoplasm [Rees MG et al. Diabetologia. 2011; Beer NL et al. Hum Mol Genet. 2009]. The resulting increase in glycolytic flux in the liver leads to reduced glucose levels and makes available substrates for de novo lipogenesis, leading to increased triglycerides. The effect of the variant is rather small, but individuals who have rare GKRP missense variants that result in a loss of function collectively have higher triglyceride levels than do people without these variants. Furthermore, an accumulation of rare GKRP variants has been reported in individuals with hypertriglyceridemia [Johansen CT et al. Nat Genet. 2010].
High-throughput assays demonstrated that a significant majority of functionally deleterious variants were collectively associated with hypertriglyceridemia and caused defects in glucokinase binding. Despite the fact that these rare variants are associated with higher triglyceride levels, they are not deterministic [Rees MG et al. Hum Mol Genet. 2014]. Prof Gloyn recommends monitoring lipid levels in phase 1 and 2 studies for GKRP small molecular disruptions (Figure 1).

GKRP-P446L Protein Has Reduced Ability to Inhibit Liver Glucokinase
Sources: Rees SD et al. Diabetologia. 2011; Beer NL et al. Hum Mol Genet. 2009.
Prof Gloyn also noted that evidence from epidemiologic, therapeutic, and genome-wide association studies demonstrates a possible link between diabetes and cancer [Voight BF et al. Nat Genet. 2010; Smith U, Gale EA. Diabetologia. 2009; Gudmundsson J et al Nat Genet. 2007]. The tumor suppressor phosphatase and tensin homolog (PTEN) antagonizes the AKT-PI3K pathway and is expressed in adipose tissue, liver, skeletal muscle, and pancreatic islets. In addition to being one of the most commonly somatically mutated genes in cancer, there are rare germline mutations in PTEN that cause the rare cancer predisposition syndrome called Cowden syndrome, which is associated with an increased risk for breast and thyroid cancer.
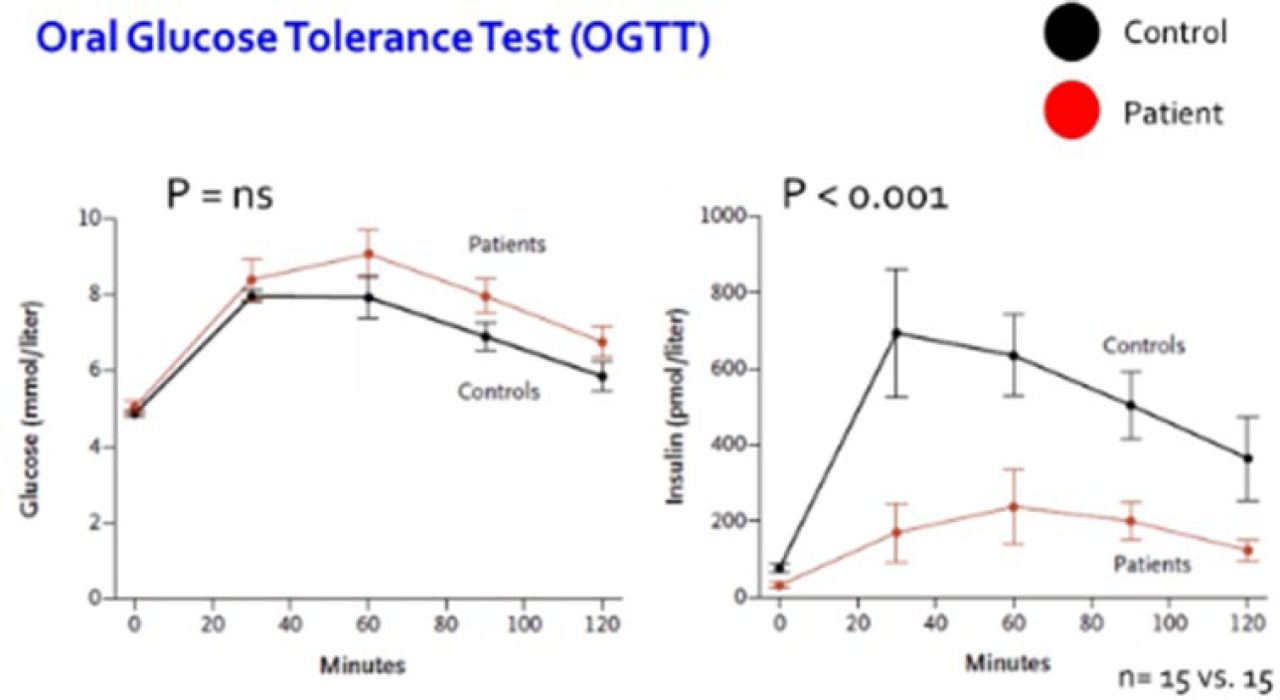
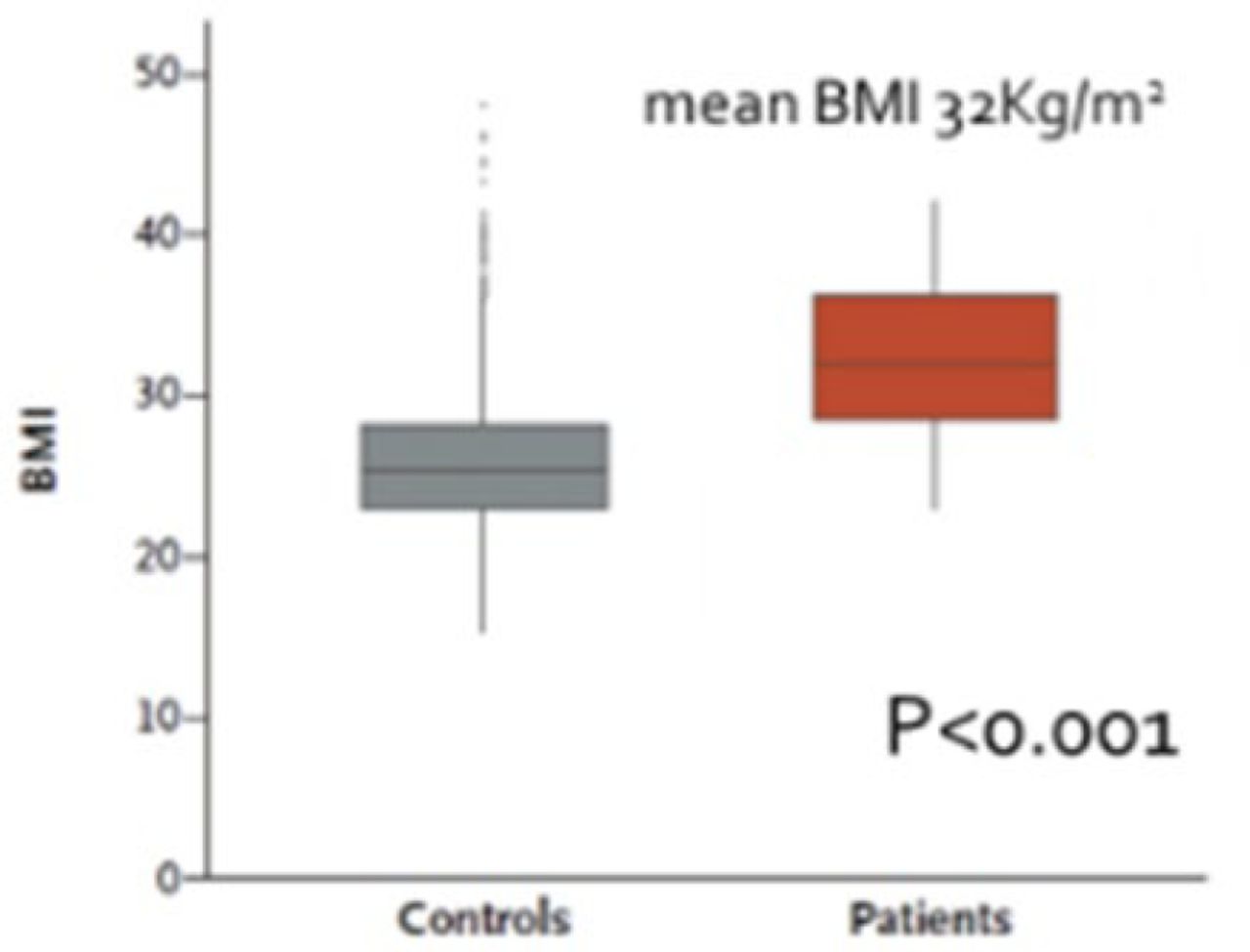
PTEN haploinsufficiency results in constitutive insulin sensitivity (Figure 2) and obesity (Figure 3). PTEN mutation carriers are more obese than population-based controls due to augmented adiposity without corresponding changes in fat distribution. The molecular mechanism for the increase in insulin sensitivity in these patients seems to be increased signaling through the AKT-PI3 kinase pathway in adipose tissue.

PTEN Mutation Causes a Decrease in Insulin Release Following an Oral Glucose Tolerance Test
PTEN, phosphatase and tensin homolog.
PTEN mutation present in patient group.
From New England Journal of Medicine, Pal A et al. PTEN Mutations as a Cause of Constitutive Insulin Sensitivity and Obesity. 2012;367:1002–1011. Copyright © 2012 Massachusetts Medical Society. Reprinted with permission from Massachusetts Medical Society.

Obesity Higher in Patients With PTEN Mutation
BMI, body mass index; PTEN, phosphatase and tensin homolog.
From New England Journal of Medicine, Pal A et al. PTEN Mutations as a Cause of Constitutive Insulin Sensitivity and Obesity. 2012;367:1002–1011. Copyright © 2012 Massachusetts Medical Society. Reprinted with permission from Massachusetts Medical Society.
In summary, Prof Gloyn said that the study of human genetics can help to identify effective therapeutic targets and determine whether the development of targeted therapies should be directed toward an agonist or antagonist. Human genetics allows us to study the effect of a perturbation on a human phenotype over the lifetime of an individual and, by doing so, to identify adverse on-target effects.

The editors would like to thank the many members of the European Association for the Study of Diabetes 2014 presenting faculty who generously gave their time to ensure the accuracy and quality of the articles in this publication.
- © 2014 MD Conference Express®
Tools









{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table of contents
Cited By...
- No citing articles found.