

Summary
This article presents a summary of the 2013 update to the 2011 American College of Rheumatology (ACR) treatment recommendations for systemic juvenile idiopathic arthritis (sJIA) [Ringold S et al. Arthritis Rheum 2013]. The purpose of the update was to expand the clinical scenarios for sJIA, to include recent data regarding the efficacy of the interleukin (IL)-1 and IL-6 inhibitors, to include macrophage activation syndrome, and to update the recommendations for follow-up tuberculosis screening for children receiving biologics.
- Rheumatological Autoimmune Disorders
- Arthritis
- Rheumatology Guidelines
- Rheumatological Autoimmune Disorders
- Rheumatology
- Arthritis
- Rheumatology Guidelines
Sarah Ringold, MD, MS, Seattle Children's, Seattle, Washington, USA, and Pamela F. Weiss, MD, MSCE, Children's Hospital of Philadelphia, Philadelphia, Pennsylvania, USA, copresented a summary of the 2013 update to the 2011 American College of Rheumatology (ACR) treatment recommendations for systemic juvenile idiopathic arthritis (sJIA) [Ringold S et al. Arthritis Rheum 2013]. The purpose of the update was to expand the clinical scenarios for sJIA, to include recent data regarding the efficacy of the interleukin (IL)-1 and IL-6 inhibitors, to include macrophage activation syndrome (MAS), and to update the recommendations for follow-up tuberculosis (TB) screening for children receiving biologics. The recommendations were developed using the RAND-UCLA Appropriateness Methodology. The primary phenotypes addressed in the update were active systemic features with varying degrees of synovitis: synovitis without systemic features, sJIA with features of concern for MAS (considered as present or absent), and repeat TB screening for all JIA patients.
For children with active sJIA and varying degrees of synovitis, depending on active joint count (AJC; 0 joints, 1 to 4 joints, >4 joints) and physician assessment of disease activity (MD global) score (<5 or ≥5), first-line therapy options include anakinra, glucocorticoid monotherapy (oral or intravenous), or nonsteroidal anti-inflammatory drugs (NSAIDs). In patients with disease activity continuing after 1 month of anakinra treatment, clinicians should consider using canakinumab, tocilizumab, methotrexate (MTX) or leflunomide, or a tumor necrosis factor-α (TNF-α) inhibitor depending on the AJC but irrespective of the MD global score. If initial therapy is glucocorticoid monotherapy and activity continues after 2 weeks of treatment, anakinra, canakinumab, tocilizumab, MTX, or leflunomide may be considered depending on MD global score. For disease activity continuing after 1 month of NSAIDs, using anakinra, glucocorticoids, canakinumab, or tocilizumab depending on MD global and AJC score may be considered. Adjunct systemic or intra-articular glucocorticoids are appropriate as needed.
Depending on the AJC, first-line therapy options when there are no active systemic features and varying degrees of synovitis include MTX or leflunomide, NSAIDs, or intra-articular glucocorticoids depending on AJC. For cases of continued disease activity after 3 months of MTX or leflunomide consider using abatacept, anakinra, TNF-α inhibitor, or tocilizumab. In patients with continued disease activity after 1 month of NSAIDs or intra-articular glucocorticoids, anakinra, MTX, or leflunomide should be considered.
For children with the third phenotype, sJIA and MAS, appropriate therapeutic options include anakinra, calcineurin inhibitors, and systemic glucocorticoids. These treatment options are not mutually exclusive.
For children in all JIA categories and those on biologics with an initial negative screen, the recommendation is for repeat TB screening at any point if the patient's risk has changed to moderate or high as determined by regional infectious disease guidelines.
Dr. Weiss noted that these phenotypes and scenarios do not address all possible sJIA presentations and that the recommendations are limited by the lack of a validated disease activity score, the low level of evidence for many recommendations, the absence of a specification of glucocorticoid dosing, route, or tapering, and the limited recommendations for MAS.
Edward M. Behrens, MD, Children's Hospital of Philadelphia, University of Pennsylvania, Philadelphia, Pennsylvania, USA, reviewed the current knowledge concerning the MAS, a systemic inflammatory response syndrome occurring in ∼10% of patients with sJIA that is characterized by coagulopathy, pan-cytopenia, hypotension, and multisystem organ failure. Labs for the condition include a falling erythrocyte sedimentation rate (ESR) and hyperferritinemia.
Although MAS is one of several cytokine storm syndromes (CSS), Dr. Behrens cautioned that clinicians should not attempt to generalize from one CSS to another. As an example, he noted while familial hemophagocytic lymphohistiocytosis (fHLH) is caused by a genetic mutation that results in disease, usually in the first year of life, MAS is associated with an underlying illness and can present at the onset or anytime during the chronic course of disease. Further, the majority of patients with MAS do not have any homozygous defects and many do not have natural killer cell killing defects. Dr. Behrens speculated that MAS may arise from some relationship with sJIA.
Occult (subclinical) MAS is identified in ∼33% of patients with sJIA who undergo bone marrow assay and may be integral to the pathogenesis of sJIA [Behrens E et al. J Rheumatol 2007]. The characteristics of subclinical MAS include a systemic inflammatory response, frequently a response of the immune system to infection, hyperferritinemia, falling ESR despite inflammation, central nervous system inflammation, and the presence of hemophagocytosis.
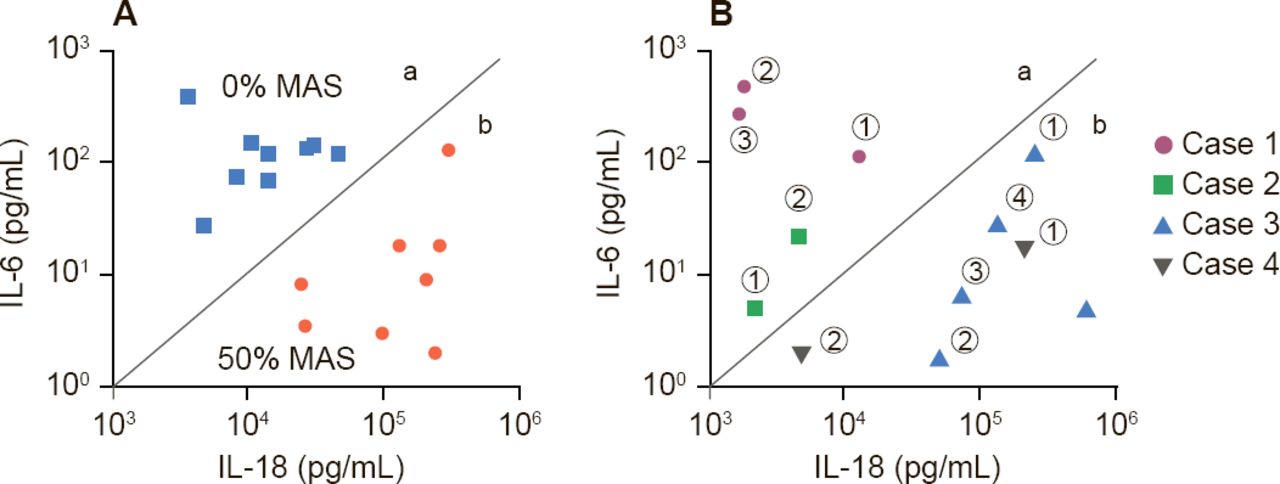
Dr. Behrens suggested two scenarios: subclinical MAS may be a spectrum of disease of sJIA, in which all patients with SJIA have some element of MAS, or there may be two types of sJIA patients—those that will get MAS and those that will not. Support for the former hypotheses comes from Fall and colleagues who identified, 225 differentially expressed genes in which cluster patterns correlated gene expression with serum ferritin levels [Arthritis Rheum 2007]. Support for the existence of two distinct types of patients may be found in a study by Shimizu and colleagues that indicates that cytokine profiling may predict subgroups of sJIA predisposed to MAS (Figure 1) [Cytokine 2013].
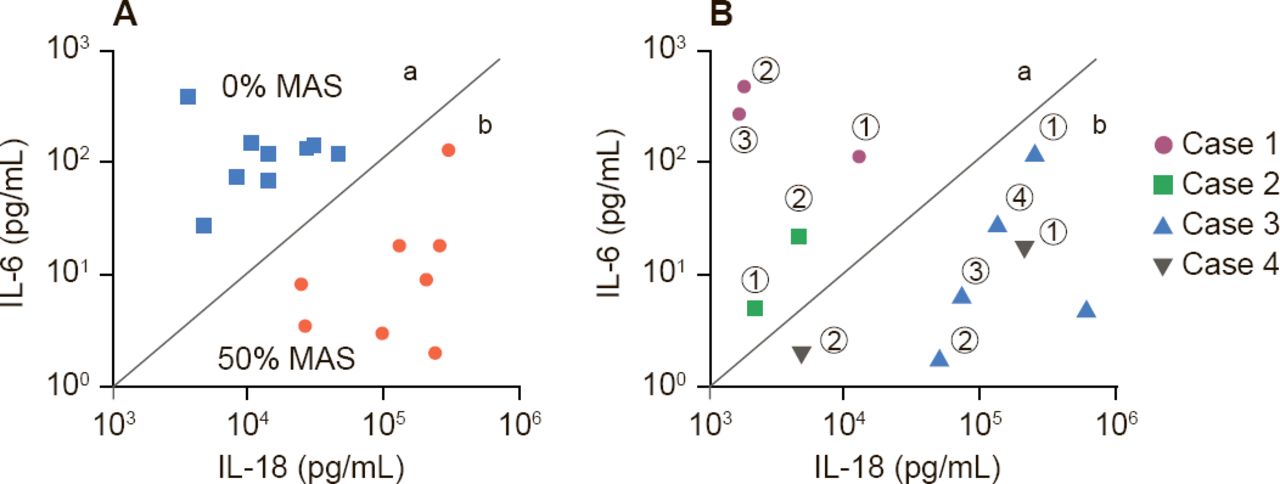
Cytokines Predict Patients With sJIA Predisposed to MAS
IL=interleukin; MAS=macrophage activation syndrome; sJIA=systemic juvenile idiopathic arthritis.
Reproduced from Shimizu M et al. Distinct subsets of patients with systemic juvenile idiopathic arthritis based on their cytokine profiles. Cytokine 2013;61(2):345–348. With permission from Elsevier.
Dr. Behrens' closing message was that sJIA and MAS represent different degrees of a common process that overlap in laboratory features, the presence of hemophagocytosis, and transcriptional program and are more common in the subset individuals with high levels of IL-18. They have a common clinical end-stage phenotype, but different mechanisms, and are likely to have different criteria for diagnosis. This information suggests the use of aggressive treatment for MAS in sJIA. IL-1 blockade, steroids, or cyclosporine A may be useful.
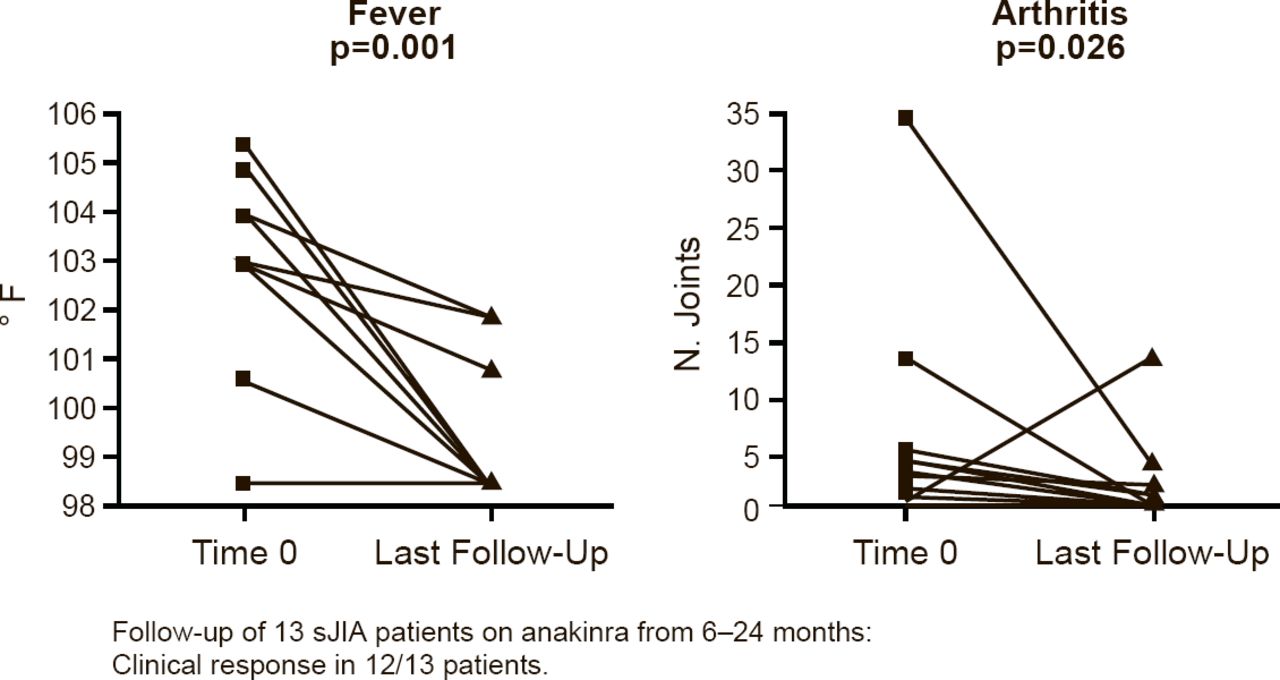
Marilynn G. Punaro, MD, University of Texas Southwestern Medical Center, Dallas, Texas, USA, presented the results of a retrospective chart review of anakinra in sJIA patients. The review comprised 13 patients who were followed for 6 to 24 months during which time 12 of the patients had a clinical response, defined as a significant decrease in fever (p=0.001) and arthritis (p=0.026; Figure 2). Significant changes were also seen in white blood cell count (p=0.004), hemoglobin (p=0.002), platelet count (p=0.001), and ESR (p=0.001).
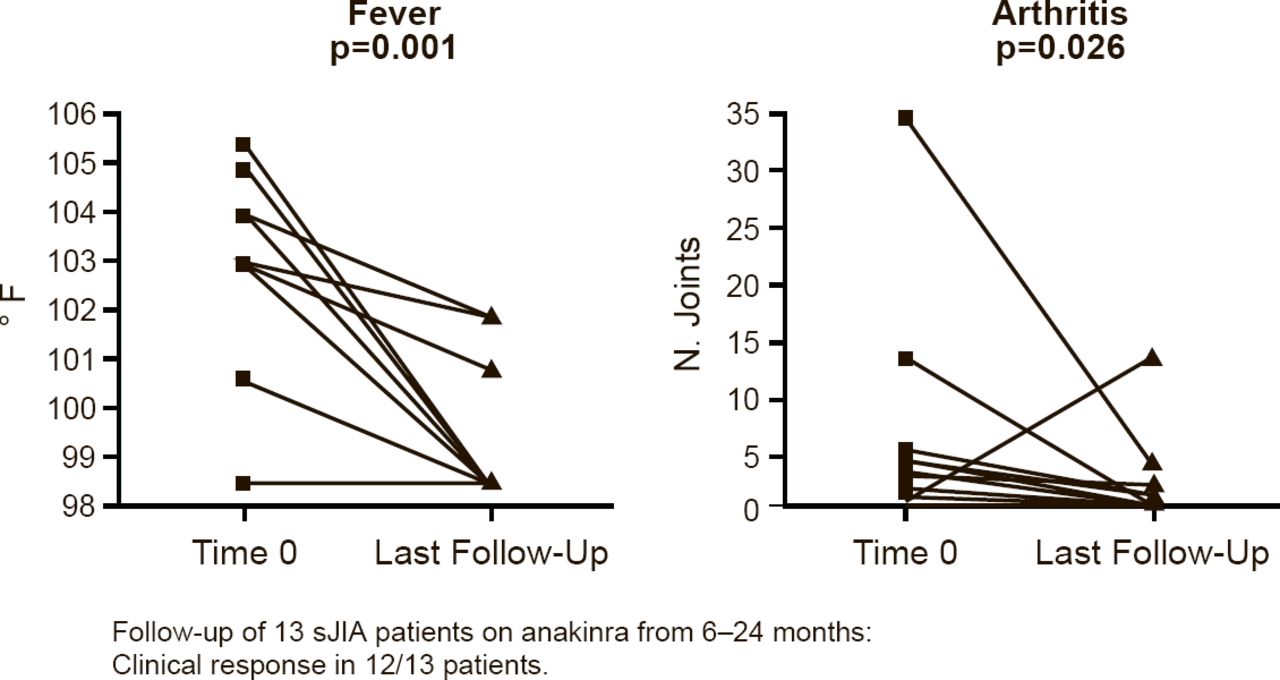
Anakinra Reduces Fever and Arthritis in Patients With sJIA
sJIA=systemic juvenile idiopathic arthritis.
Reproduced with permission from MG Punaro, MD.
Dr. Punaro then reviewed unpublished data from the 51 children (including the original 13) with a blood gene-expression profile compatible with sJIA who were treated with anakinra (2.67 mg/kg SC QD) for at least 1 week at her institution. After a median of 5 years of follow-up, 38 patients (75%) had inactive disease while clinical remission (on medication) was noted in 31 patients (60%). Eight patients (16%) patients had a partial response and 5 (10%) had no response. Minor complications associated with anakinra treatment included pain, burning with injection, injection-site reaction, infections, and influenza. There was one death and four patients developed MAS while on anakinra. MAS patients responded to treatment with increased anakinra and added steroids. Patients flaring after discontinuation of anakinra may not be responsive on re-treatment.
Dr. Punaro said that the IL-1 receptor antagonist, anakinra, is an effective, safe, and well-tolerated treatment for sJIA.
- © 2013 MD Conference Express®
Tools









{kind=link}
{kind=link}
Table of contents
Cited By...
- No citing articles found.