

Summary
Molecular research over the past 25 years has yielded a wealth of data on the molecular and genetic features of von Willebrand disease. This article discusses the translation of this molecular knowledge into practical aspects of clinical diagnosis and treatment of this bleeding disorder. Also discussed is the future of coagulation factor replacement therapy for hemophilia.
- Hemorrhagic Disorders
- Hemorrhagic Disorders
- Hematology
TRANSLATIONAL MEDICINE ADVANCES IN VON WILLEBRAND DISEASE
Molecular research over the past 25 years has yielded a wealth of data on the molecular and genetic features of von Willebrand disease (VWD). David Lillicrap, MD, Queen's University Kingston, Ontario, Canada, discussed the translation of this molecular knowledge into practical aspects of clinical diagnosis and treatment of this bleeding disorder.
The gene for human von Willebrand factor (VWF) was cloned and characterized by four groups in 1985 [Verweij CL et al. Nucleic Acids Res 1985; Ginsburg D et al. Science 1985; Sadler IE et al. Proc Natl Acad Sci USA 1985; Lynch DC et al. Cell 1985]. Located on the short arm of chromosome 12, the VWF gene comprises 178 kilobases of DNA and 52 exons. Genetic analysis of this complex gene can be difficult due to the presence of a pseudogene sequence on chromosome 22 that differs in only 3% of the sequence analysis compared to the sequence in the middle of the VWF gene. Annotation of the VWF protein structure has recently been revised, with the D domains now referred to as D assemblies composed of smaller molecules, including von Willebrand D (VWD), 8-cysteine (C8), trypsin inhibitor-like (TIL), and E domains [Zhou YF et al. Blood 2012]. The former B and C regions of the protein are now annotated as 6 tandem VWC and VWC-like domains [Zhou YF et al. Blood 2012], which provide length and flexibility under conditions of shear.
The diagnosis of VWD involves three main components: a personal history of excessive mucocutaneous bleeding; laboratory hemostasis results compatible with quantitative or qualitative VWF pathology; and a family history of VWD. Mucocutaneous bleeding is evaluated using bleeding assessment tools such as the International Society on Thrombosis and Haemostasis (ISTH) -Bleeding Assessment Tool (BAT) [Rydz N, lames PD. J Thromb Haemost 2012]. According to Prof. Lillicrap, laboratory hemostasis testing has not changed much in the last 20 years. Hemostasis assays include those for VWF antigen (VWF:Ag); ristocetin cofactor (VWF:RCo); factor VIII binding (VWF:FVIIIB); collagen binding (VWF:CB); VWF propeptide (VWFpp) used as surrogate for clearance rate of VWF); multimeric analysis (VWF multimers); and ristocetin platelet agglutination (RIPA).
Genetic analysis is useful for family planning and management of VWD. The University of Sheffield, United Kingdom, is home to a VWD mutation database [www.vwf.group.shef.ac.uk] that currently describes 399 unique VWF gene mutations. VWF mutation detection rates are −65% in type 1,90% in type 2 (2A, 2B, 2M, and 2N), and 85% in type 3 VWD. Currently, the number of known mutations is 127 in type 1, 75 in type 2A, 25 in type 2B, 29 in type 2M, 31 in type 2N, and 112 in type 3. The VWF genetic locus contains 181 polymorphisms, adding to the complexity of diagnosis and management.
In a study of VWD mutation patterns, Yadegari H et al. [Thromb Haemost 2012] found mutations in 68%, 94%, and 94% of VWD type 1, 2, and 3, respectively. Of the mutations found in all VWD types, 54.4% were missense mutations, 14.7% were small deletions, 2.9% were small insertions, 10.3% were nonsense mutations, 7.4% were splice-site mutations, 8.8% were large deletions, and 1.5% were silent mutations. In patients with type 2 VWD, genetic testing can differentiate between identical phenotypes caused by mutations in different genes. For example, type 2B and platelet-type VWD have the same phenotypes but are due to mutations of the VWF and GP1ba genes, respectively. This distinction is important for selecting therapy (VWF and FVIII concentrate for VWF vs platelets for platelet-type).
The CHARGE study identified 5 novel candidate genetic loci strongly associated with levels of VWF [Smith NL et al. Circulation 2010]. Syntax binding protein 5 (STXBP5) and syntaxin 2 (STX2) are associated with vesicular trafficking and exocytosis. Scavenger receptor class A member 5 (SCARA5), stabilin 2 (STAB2), and C-type lectin type 4 member M (CLEC4M) are receptor proteins that might be involved in clearing VWF.
Few advances have been made in the treatment of VWD over the past 20 years. Desmopressin is effective but therapeutic trials including 1-, 2-, and 4-hour time points (to check for accelerated clearance) are required. Plasma derived (pd) concentrates are also available. The pd-VWF/FVIII concentrates ranges from very high to intermediate purity and have variable RCo/Ag and RCo/FVIII levels (Table 1). Although these concentrates are safe and effective, their optimal use is not fully understood.
Plasma-Derived VWF/FVIII Concentrates Licensed in Europe
A recent study of the safety, tolerability, and pharmacokinetics of a Chinese hamster ovary (CHO) cell-derived rVWF (VWF:RCo/FVIII:C ratio 1.3:1) in 32 patients showed that it is safe and the pharmacokinetics of rVWF:RCo activity, VWF:Ag, and collagen binding activity (VWF:CB) were similar to those of pd-VWF/FVIII [Mannucci PM et al. Blood 2013].
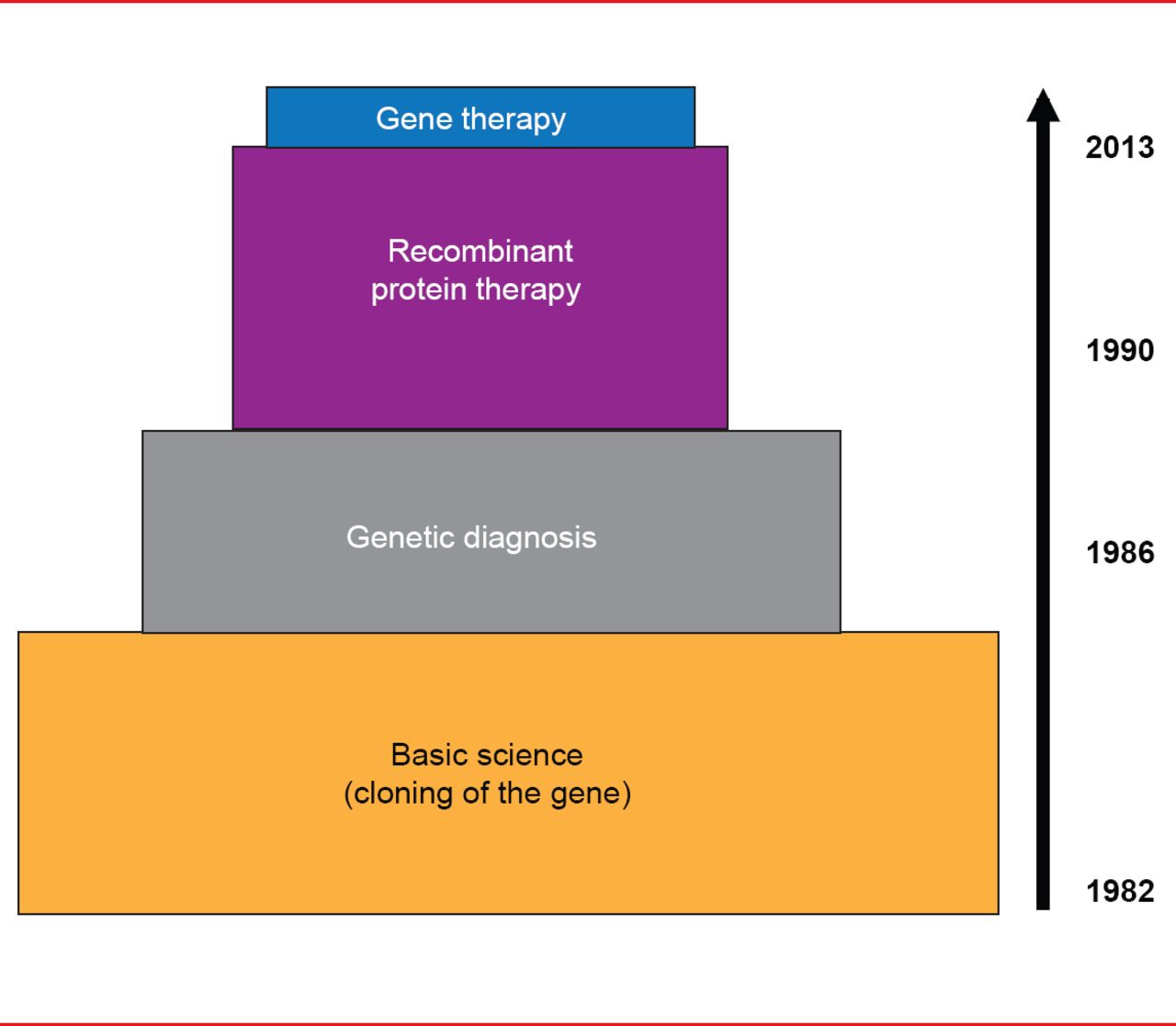
Translational medicine progress has been much more rapid in hemophilia versus VWD. Cloning of the FVIII and FIX genes in the early 1980s led to rapid use of genetic diagnosis for hemophilia in the mid-1980s and recombinant protein therapy in the 1990s, and finally, to gene therapy for hemophilia B in 2012 (Figure 1).

Translational Medicine Advances in Hemophilia
Reproduced with permission from D Lillicrap, MD.
Translational progress in VWD has been slower, with extensive basic science research ongoing since cloning of the gene in 1985. Genetic diagnosis was only introduced in the past decade, while research on recombinant protein therapy and gene therapy began just a few years ago (Figure 2).

Translational Medicine Advances in von Willebrand Disease
Reproduced with permission from D Lillicrap, MD.
The relatively slow progress in translational advances in VWD is due to a number of factors. First, bleeding symptoms in type 1 VWD are mild compared with those in hemophilia. In the past, women's bleeding problems with VWD have not been addressed, although this is changing. Additionally, phenotypic testing for type 2 and 3 VWD is straightforward, resulting in less need for genetic testing. Although phenotypic diagnosis of type 1 VWD is difficult, genetic diagnosis also is difficult. Other reasons include the complexity of VWD genetics, the safety and efficacy of desmopressin and pd-concentrates, and the rarity of VWF immunogenicity. Prof. Lillicrap noted that although the translational clinical potential of research in VWD has not been achieved, the biologic potential is enormous, considering the many functions of VWF including its contributions to venous thrombosis, atherothrombosis, platelet adhesion and aggregation, inflammation, angiogenesis, cell proliferation, and apoptosis.
FUTURE OF COAGULATION FACTOR REPLACEMENT THERAPY FOR HEMOPHILIA
Substantial improvements in hemophilia care have been achieved during the past 30 years. The current treatment standard is intravenous (IV) replacement therapy, given as needed during bleeding episodes or prophylactically on a routine schedule. Although safe and highly effective, hemophilia therapy involves frequent injections due to a short half-life and is immunogenic, costly, and unavailable to 70% of patients worldwide. These issues were addressed by Flora Peyvandi, MD, PhD, University of Milan, Milan, Italy, in her presentation on new generation therapies for hemophilia.
Strategies to extend the half-life, increase efficacy, and reduce immunogenicity of hemophilia therapies are under investigation, including the addition of polyethylene glycol (PEG) polymers and polysialic acids, drugs formulated with PEG-modified liposomes, and bioengineered fusion protein therapies. The covalent coupling of PEG to a protein product, known as pegylation, increases the drug's dynamic volume, resulting in reduced clearance by the kidneys and greater efficacy. Pegylation also masks the protein surface, which reduces immunogenicity and proteolytic degradation. This technology has been applied to FVIII, FIX, and FVIIa.
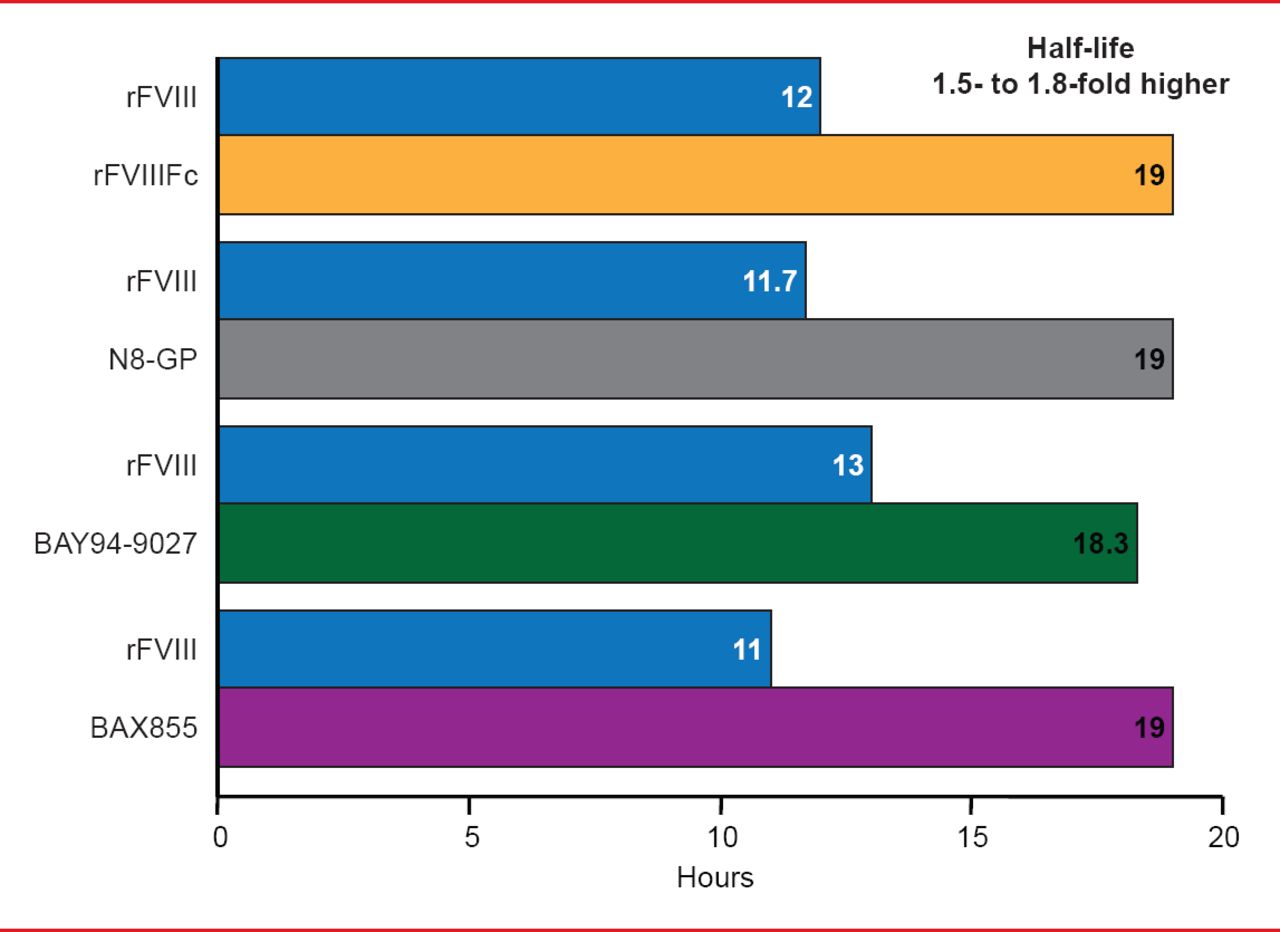
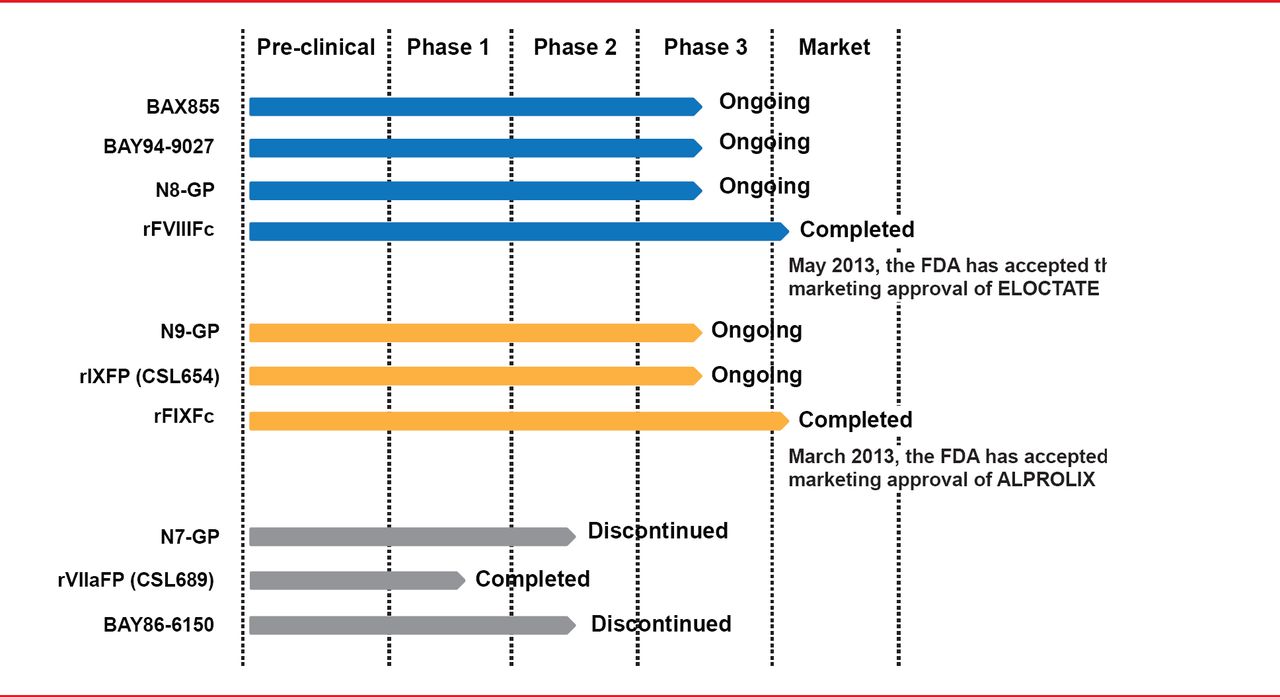
Targeted pegylation is a technique in which PEG is attached to specific regions of coagulation factors. Glycopegylation of FVIII involves attaching PEG to the O-linked glycan in the truncated B-domain of rFVIII. Thrombin-mediated activation of the pegylated molecule releases the PEG B-domain, resulting in a fully functional activated molecule similar to endogenous FVIIIa [Stennicke HR et al. Blood 2013]. In another technique, the Fc fragment of immunoglobulin or albumin is fused to the coagulation factor, increasing reuptake and half-life. Four long-acting rFVIII products are in clinical trials, including 3 ongoing studies and 1 completed study (Table 2). The half-lives of these new products have been extended 1.5- to 1.8-times the half-life of rFVIII (Figure 3). Prophylactic treatment with long acting rFVIII will involve 1 to 2 infusions per week compared with 2 to 3 per week with standard rFVIII.
Clinical Studies of Long-Acting rFVIII

Half-Life Extension of Long Acting rFVIII Products
Reproduced with permission from F Peyvandi, MD, PhD.
Three long acting rFIX products are being studied in Phase 2 and 3 trials (Table 3). The half-lives of these products are extended 3- to 5-times the half-life of standard rFIX (Figure 4). Prophylaxis with long acting rFIX will require infusions every 1 or 2 weeks compared with 2 weekly infusions of standard rFIX.
Clinical Studies of Long Acting rFIX

Half-Life Extension With Long-Acting rFIX Products
Reproduced with permission from F Peyvandi, MD, PhD.
Three long-acting rFVIIa products have been studied but two were discontinued due to unsatisfactory dose-response (N7-GP) and immunogenicity (BAY86-6150). A Phase 1 trial has been successfully completed for the remaining product, CSL689, an albumin fusion rFVIIa with a half-life 3- to 4-times longer than standard rFVIIa [NCT01542619].
Pegylated proteins are well tolerated but vacuolization of renal tubules has been observed in animal models. It is possible that renally cleared pegylated proteins or their metabolites accumulate in the kidneys, causing formation of PEG hydrates that interfere with glomerular filtration. According to Prof. Peyvandi, long-term safety evaluations of these products are needed because data from prelicensing studies are insufficient to demonstrate their safety. Polypeptide chains are biologic alternatives to pegylation that are stable in plasma, fully biodegradable, do not accumulate in organs, and are not toxic or immunogenic in animals. Examples of these are half-life extension technology (XTEN), PASylation (PAS), elastin-like polypeptide (ELP) technology, and HESylation (HES).
Several alternative therapeutic strategies have been under investigation, including inhibition of tissue factor pathway inhibitor (TFPI; BAX499), inhibition of antithrombin(ALN-AT3), and bispecific antibody(ACE910). Studies of the TFPI inhibitor, BAX499 have stopped due to increased bleeding events.
The antithrombin inhibitor, ALN-AT3, was developed using RNA interference (RNAi). RNAi involves gene silencing in a sequence-specific manner at the mRNA level. The mechanism involves breaking a double-stranded RNA (dsRNA) that matches a specific gene sequence into short pieces of RNA called small interfering RNAs (siRNAs) that can target a specific gene sequence. ALN-AT3 is an siRNA that targets antithrombin in the liver using a hepatocyte targeting ligand. Preclinical studies show that ALN-AT3 inhibits antithrombin in nonhuman primates. Antithrombin reduction was associated with increased thrombin generation and up to 4-fold increases in peak thrombin. ALN-AT3 has a long duration of action with the potential for once weekly or twice monthly subcutaneous dosing.
The bispecific antibody, hBS23/ACE910 has the same activity as FVIII, binding to both FIXa and FX and increasing thrombin generation [Kitazawa T et al. Nat Med 2012]. IV hBS23/ACE910 has a half-life of 2 weeks, while bioavailability of subcutaneous hBS23/ACE910 is 84%. A Phase I clinical study of this drug currently is recruiting in Japan [JapicCTI-121934].
Considerable progress has been made in the treatment of hemophilia over the past 25 years but few options are available for patients with rare bleeding disorders. Fresh frozen plasma and cryoprecipitate are still used in most countries because specific products for specific deficiencies are not available everywhere. Novel drugs for the treatment of hemophilia and other bleeding disorders have the potential to change patients' lives but many issues remain. Among these are defining novel drugs for orphan drug status, the market impact of biosimilar drugs, and drug registration differences in different countries. Cost and reimbursement issues for novel drugs can present obstacles to global availability. Despite these issues, gene-and protein-based research is ongoing, with the goal of bringing a cure for bleeding disorders one step closer to reality, concluded Prof. Peyvandi.

Current Status of Long Acting Coagulation Factor Products.
Reproduced with permission from F Peyvandi, MD, PhD.
- © 2013 MD Conference Express®
Tools









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table of contents
Cited By...
- No citing articles found.