

Summary
In the 44th Claude Bernard Lecture on diabetes, Daniel J. Drucker, MD, University of Toronto, Toronto, Ontario, Canada, discussed his research findings concerning incretin hormone action and the treatment of diabetes. The lecture covered the pharmacological/physiological mechanisms of the glucagon-like peptides GLP-1 and GLP-2, and their relevance to the treatment of human disease.
- Infectious Disease
- Hormone Therapy
- Diabetes Mellitus Diabetes & Metabolic Syndrome
In the 44th Claude Bernard Lecture on diabetes, Daniel J. Drucker, MD, University of Toronto, Toronto, Ontario, Canada, discussed his research findings concerning incretin hormone action and the treatment of diabetes. The lecture covered the pharmacological/physiological mechanisms of the glucagon-like peptides GLP-1 and GLP-2, and their relevance to the treatment of human disease.
The importance of GLP-1 system basal control of glucose homeostasis was first evidenced in animal studies in which GLP-1 receptor (GLP-1R) knockout (−/−) mice exhibited increases in fasting blood glucose and decreases in circulating insulin following oral and intraperitoneal glucose challenges [Scrocchi LA et al. Nat Med 1996]. Since then, numerous studies have shown that the GLP-1/GLP-1R system, although classically focused on the β-cell, is also critically important for the physiology of many other systems (eg, energy expenditure, central nervous system stress response, and the immune system).
Given that glucagon is a critical hormone in the pathophysiology of diabetes and the control of glucose homeostasis, can hyperglucagonemia of type 2 diabetes mellitus (T2DM) be attenuated successfully and used as a treatment for diabetes? One concern is whether blocking the glucagon receptor (Gcgr) is the right approach. GLP-1 and GLP-2 play key roles in cell proliferation and survival, and it is possible that the Gcgr may have a similar function that would make blocking it a poor choice for T2DM therapy.
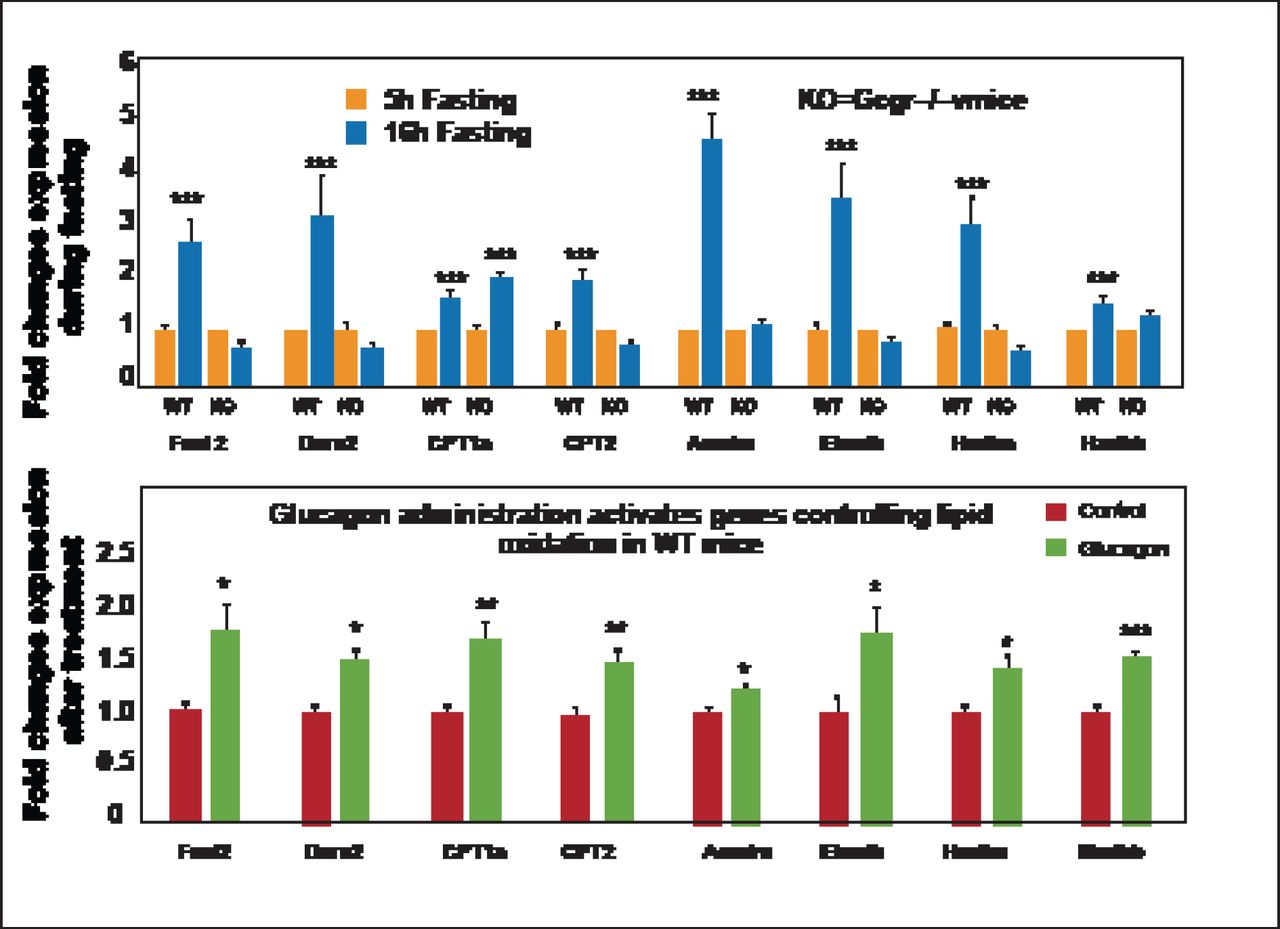
Gcgr−/− mice have shown that the Gcgr is required for control of lipid metabolism during the adaptive metabolic response to fasting. Glucagon inhibits hepatic lipid production and acutely increases hepatic lipid secretion. Fasting Gcgr−/− mice are unable to control plasma lipids and experience an increase in triglyceride secretion from the liver. These mice are also resistant to diet-induced obesity and fat deposition in peripheral organs; however, they do not resist lipid deposition in the liver, somewhat negating any therapeutic potential [Longuet C et al. Cell Metab 2008]. Gcgr−/− mice also show enhanced susceptibility to hepatosteatosis on a methionine choline-deficient diet. Restoration of hepatic Gcgr expression attenuates the development of hepatocellular injury, which extends the essential actions of the Gcgr beyond the metabolic control of glucose homeostasis to encompass the regulation of hepatocyte survival [Sinclair EM et al. Gastroenterology 2008]. Independent of the lipid pathway, Gcgr plays a critical role in the balance between survival and cell death by way of lipid oxidation control. During fasting, increases in many of the genes in the liver important for liver oxidation are seen, but Gcgr−/− mice are unable to activate similar hepatic gene expression systems (Figure 1) [Longuet C et al. Cell Metab 2008]. Thus, glucagon actions in the liver are essential for both hepatocyte survival and lipid accumulation.

Ggcr Signaling Is Essential for Control of Hepatic Lipid Oxidation.
* p<0.05; ** p < 0.01; *** p<0.001. KO=knockout; WT=wild-type. Reprinted from Longuet C et al. The Glucagon Receptor Is Required for the Adaptive Metabolic Response to Fasting. Cell Metab Nov 5, 2008;8(5):359–371, with permission from Elsevier.
Gcgr also plays an important role in the pancreas in that Gcgr−/− mice have increased pancreatic weight, enhanced β-cell function, and massive islet and α-cell hyperplasia. Reductions in Gcgr signaling direct the pancreas to enhance islet α-cell proliferation, which develops independently of GLP-1R in Gcgr−/− mice [Ali S et al. J Clin Invest 2011]. The liver sends the signal to β-cells to proliferate; in control experiments genetic disruption of liver Gcgr sends a signal promoting proliferation of transplanted wild-type (WT) α-cells under the kidney capsule. But α-cell hyperplasia and proliferation can still occur very rapidly if WT islets are placed under the kidney capsule in mice with liver-selective inactivation of Gcgr. Thus, organized nerves, α-cells within the islets, or even islet cells organized within their normal location are not needed for this signal to be communicated [Longuet C et al. Diabetes. In press]. The importance of liver Gcgr to the α-cell axis has also been shown in humans. In a recent clinical study the Gcgr antagonist MK-0893 produced robust reductions in HbA1C levels in patients with T2DM [Engel SS et al. ADA 2011 Abstract 309-OR] but was associated with elevations in transaminase and lipids.
The attenuation of Gcgr signaling produces substantial improvement of glucose homeostasis in mice and humans. This signaling is essential for hepatocyte survival, lipid metabolism, and controlling the rate of α-cell proliferation—likely through a circulating factor.
However, the therapeutic window for reduction of glucagon action to manifest beneficial effects for glucose control while avoiding enhancement of hepatic lipid storage, dyslipidemia, hepatocyte injury, and α-cell proliferation in diabetic subjects is unclear.
Two classes of medication that enhance incretin action (the GLP-1R agonists and dipeptidyl peptidase-4 [DPP-4] inhibitors) are used to treat T2DM. Prof. Drucker discussed the cardiovascular biology of these medications, including their direct and indirect effects on cardiomyocytes, blood vessels, adipocytes, blood pressure control, and postprandial lipoprotein secretion.
When the mesenteric arteries of WT and Glp1r−/− mice were treated with GLP-1, GLP-1 (9–36), and exendin-4 (a GLP-1 agonist), GLP-1 and GLP-1 (9–36) exerted direct vasodilatory effects but exendin-4 did not. When the degradation of native GLP-1 is blocked by the addition of a DPP-4 inhibitor, vasodilation is substantially reduced but not completely eliminated in either group. This is likely because GLP-1 (9–36), a more potent vasodilator than GLP-1, was acting directly on the blood vessel [Ban K et al. Circulation 2008]. It has been suggested that there are GLP-1 receptors on endothelial or smooth muscle cells. However, in preconstricted mouse arteries the degradation-resistant GLP-1R agonist liraglutide does not directly vasodilate these arteries, but acetylcholine does. Support for the hypothesis that there is no direct action on blood vessels for these GLP-1R agonists comes from a recent clinical trial that showed exenatide BID for 3 months had no effect on microvascular endothelial function in obese, nondiabetic subjects [Kelly AS et al. Cardiovasc 2012]. In the cardiovascular system, pretreatment with liraglutide before coronary artery occlusion in normal and diabetic mice improved survival, reduced cardiac rupture and infarct size, and improved cardiac output. Liraglutide also modulated the expression and activity of cardioprotective genes in the mouse heart in a GLP-1R-dependent manner [Noyan-Ashraf MH et al. Diabetes 2009].
GLP-1R is expressed in the cardiovascular system in the heart and blood vessels, and, although activation of the GLP-1R is cardioprotective, the mechanisms by which this occurs are poorly understood and most likely the result of both direct and indirect action. Activation of GLP-1R also modulates inflammation; however, GLP-1R is not expressed in most mouse macrophage populations and further analyses requires a more stringent methodology.
GLP-1 is synthesized in the enteroendocrine L cells and in the islet α-cells in the pancreas; however, nutrients increase L cell GLP-1 secretion and proglucagon-derived peptide biosynthesis but decrease glucagon biosynthesis and secretion in the islet α-cell. The molecular basis for this differential biology can be detected in the gene expression profiling of these two entities. Progesterone (P4) is an unexpected modulator of GLP-1 secretion in mice: enteral P4 increases plasma levels of GLP-1 and enhances glucose tolerance independent of the classical progesterone. The membrane progesterone receptors Paqr5 and Paqr7 are expressed in the gastrointestinal tract and in gut endocrine cell lines, and are essential for P4-mediatged GLP-1 secretion in GLUTag cells. Gut-restricted Paqr5 and Paqr7 agonists may enhance incretin secretion and control glucose homeostasis without systemic absorption of the ligands.
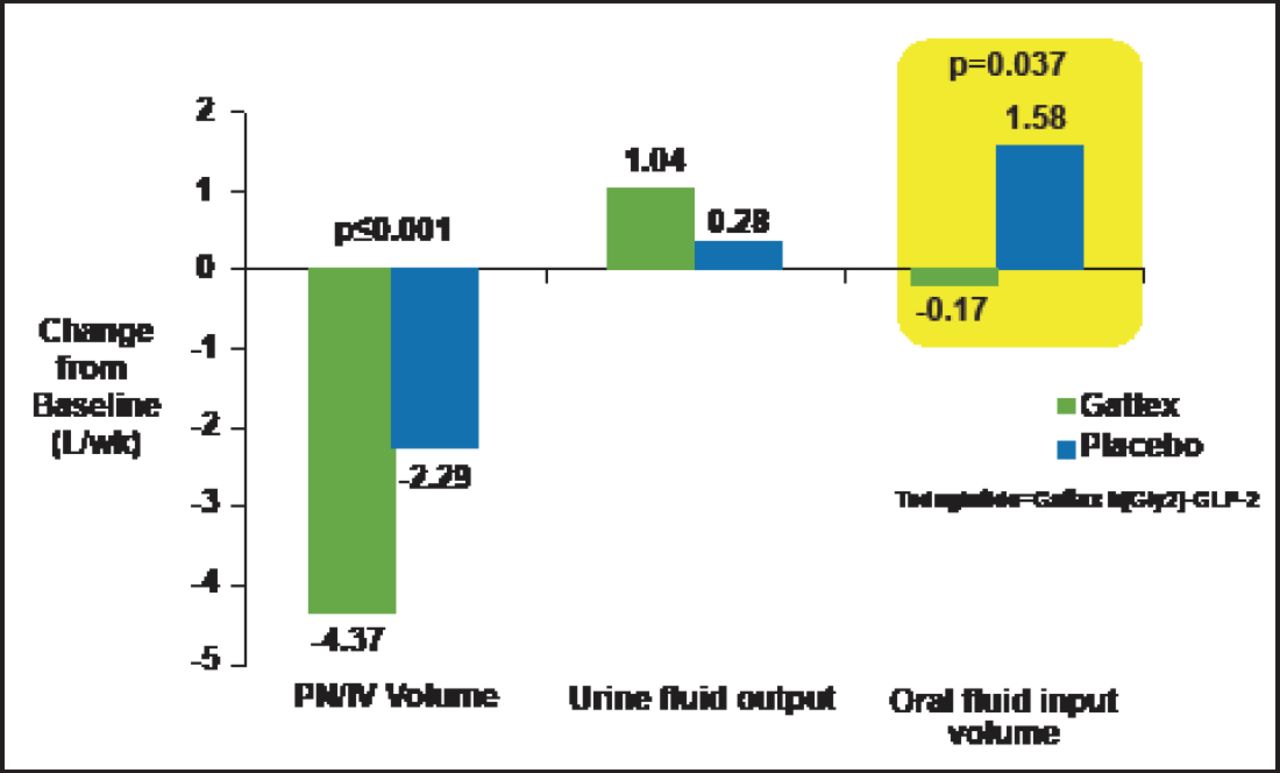
GLP-2, a 33-aa peptide, stimulates crypt cell proliferation and produces a marked increase in bowel weight and villus growth of the jejunum and ileum. Glp2r-/- mice exhibit enhanced sensitivity to nonsteroidal anti-inflammatory drug-induced gut injury and abnormal host-bacterial interactions [Lee SJ et al. Endocrinology 2012]. GLP-2 also has a role in the control of nutrient absorption. GLP-2 rapidly enhances intestinal blood flow; increases the absorption of sugars, proteins, and fats; reduces acid secretion; and delays gut motility. Teduglutide, a GLP-2 analogue, reduced volume and number of days of parenteral support for patients with short bowel syndrome, suggesting GLP-2 can restore nutritional balance and reduce parenteral nutrition requirements in these subjects (Figure 2) [Jeppesen PB et al. Gastroenterology 2012].

Teduglutide Reduces Parenteral Nutrition Requirements in Human Subjects with Short Bowel Syndrome.
Reprinted from Jeppesen PB et al. Teduglutide Reduces Need for Parenteral Support Among Patients With Short Bowel Syndrome With Intestinal Failure. Gastroenterology Jan 1, 2012; S0016–5085(12) 01316–9, with permission from Elsevier.
GLP-1 has many intriguing effects on blood vessels and the heart that are of interest to clinicians. Prof. Drucker concluded by saying, “What I've tried to illustrate is that the techniques we use to detect GLP-1R expression either at the RNA [ribonucleic acid] level or protein level perhaps need a little bit more thought and rigor in terms of interpretation. And finally I've tried to convince you that perhaps GLP-2…may potentially be useful in the treatment of patients who have trouble with their own endogenous gut functions.” Considerable progress has been made, but considering the number of patients with diabetes and the advanced technology available, there is still a lot of work to do in the treatment of patients with diabetes.
- © 2012 MD Conference Express®
Tools









{kind=link}
{kind=link}
Table of contents
Cited By...
- No citing articles found.