

Summary
Current treatment of hyperglycemia in type 2 diabetes mellitus is often ineffective and has unwanted side effects. Therefore, novel antidiabetic drugs are under development to address this problem. Sodium glucose cotransporter 2 inhibitors are currently being tested for safety and efficacy [DeFronzo R et al. Endcorinology Rev 2011].
- Diabetes Mellitus
Current treatment of hyperglycemia in type 2 diabetes mellitus (T2DM) is often ineffective and has unwanted side effects. Therefore, novel antidiabetic drugs are under development to address this problem. Sodium glucose cotransporter 2 (SGLT2) inhibitors are currently being tested for safety and efficacy [DeFronzo R et al. Endcorinology Rev 2011]. Ernest M. Wright, DSc, David Geffen School of Medicine, UCLA, Los Angeles, California, USA, discussed the SGLT gene family, the molecular dynamics of Na+ glucose cotransport, genetic disorders of SGLTs, and the mechanism of action of SGLT2 inhibitors.
There are two classes of glucose transporters that are involved in glucose homeostasis in the body—the facilitated transporters, or uniporters (GLUTs), and the active transporters, or symporters (SGLTs). The energy for active glucose transport is provided by the sodium gradient across the cell membrane, the Na(+) glucose cotransport hypothesis first proposed in 1960 by Crane [Wright EM et al. Physiol Rev 2011].
The human SGLT (SLC5) gene family is made up of 12 members. They belong to a structural class of membrane proteins from unrelated gene families of antiporters and Na(+) and H(+) symporters [Wright EM et al. Physiol Rev 2011]. Membrane cotransport proteins that use a five-helix inverted repeat motif have emerged as one of the largest structural classes of secondary active transporters [Watanabe et al. Nature 2010].
Watanabe et al. identified a cascade of events that were initiated by sodium release that ensure proper timing of ion and substrate release. Once set in motion, these molecules weaken substrate binding to the transporter and allow galactase to readily enter the intracellular space [Watanabe A et al. Nature 2010].
The human Na(+)/D-glucose cotransporter (hSGLT2) is believed to be responsible for the bulk of glucose reabsorption in the kidney proximal convoluted tubule. Since blocking reabsorption increases urinary glucose excretion, hSGLT2 has become a novel drug target for T2DM treatment. Results from hSGLT1 research suggest that it may also play a significant role in the reabsorption of filtered glucose in the late proximal tubule [Hummel CS et al. Am J Physiol Cell Physiol 2011]. According to Dr. Wright, SGLT1 is essential for normal intestinal absorption of glucose.
The kidney contributes to glucose homeostasis through gluconeogenesis and the use and reabsorption of glucose from the glomerular filtrate [Gerich JE. Diabet Med 2010]. Dr. Wright noted that diabetes is a disorder of glucose homeostasis. One of the earliest symptoms is a loss of glucose to urine due to hyperglycemia, overwhelming the reabsorption capacity of SGLTs in the proximal tubule. There is a growing interest in alternative therapies to manage diabetic patients, and one is to control blood glucose by inhibiting SGLT2 in the kidney.
The pharmaceutical industry has been encouraged by 1) studies that have demonstrated that intravenous phlorizin decreases blood glucose levels without producing hypoglycemia in diabetic animals and that this is accompanied by an improvement in insulin resistance; 2) reports that familial renal glucosuria is a benign disorder with no long-term renal abnormalities; and 3) cloning of the “renal” SGLTs, enabling in vitro studies [Wright et al. Physiol Rev 2011].
The strategy has been to develop oral SGLT2 inhibitors. Oral phlorizin does not fit the bill, as it blocks gastrointestinal absorption of glucose and thus produces osmotic diarrhea. Furthermore, phlorizin is hydrolyzed to glucose and phloretin by the intestinal brush border lactase-hydrolase; so, little of the intact molecule is absorbed.
Yet, most SGLT2 inhibitors in the drug pipeline have exploited the same chemical space as phlorizin, and many of these compounds are in Phase 1 to Phase 3 clinical trials. Several SGLT2 inhibitors are undergoing development, including dapagliflozin, canagliflozin, ASP1941, LX4211, and B1I10773 [Chao EC, Henry RR. Nat Rev Drug Discov 2010]. Dapagliflozin reduces fasting and postprandial plasma concentrations of glucose and HbA1C, as well as body weight, with low risk of hypoglycemia [Tahrani AA et al. Lancet 2011].
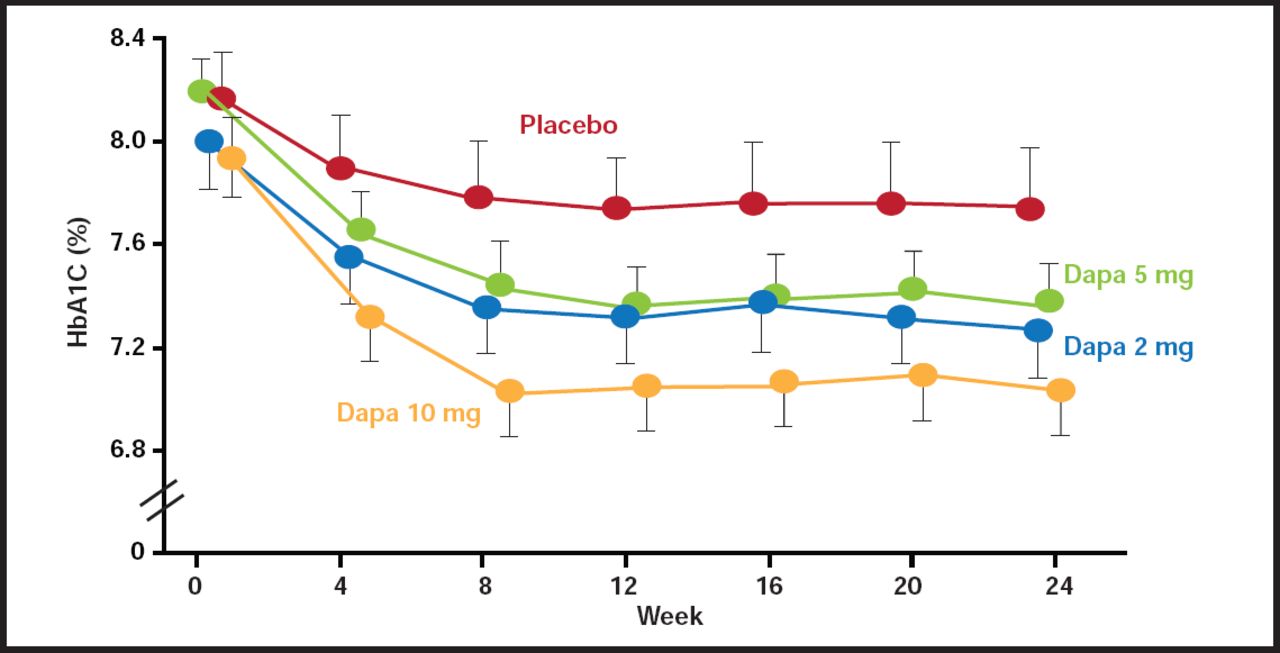
Safety studies of dapagliflozin in healthy human subjects indicate that the drug is well tolerated, with minimal adverse events and no clinically relevant changes in vital signs relative to the placebo controls. The drug is currently under review by the Food and Drug Administration for treatment of diabetes (Figure 1).

Time Course of Change in HbA1C in Dapagliflozin-Treated Diabetic Subjects.
Reproduced with permission from from the Lancet, from “Management of type 2 diabetes: new and future developments in treatment”; Tahrani A et al, vol. 378, no. 9786, 182, 2010; permission conveyed through Copyright Clearance Center, Inc.
Dr. Wright concluded his presentation by making six key points about SGLTs:
-
Those in the small intestine and kidney are drug targets for diabetes
-
SGLT2 in the kidney is the major focus of pharmaceutical companies
-
They are not essential for life (as shown by glucose-galactose-malabsorption and familial renal glucosuria)
-
Selective SGLT2 inhibitors have been developed
-
SGLT2 inhibitors improve glucose homeostasis in diabetic rats
-
SGLT2 inhibitors in Phase 3 trials have had promising results and no remarkable adverse events
Drugs Targeting the Metabolic Syndrome: Selective PPAR Agonists
The peroxisome proliferator-activated receptors (PPARs) and the retinoid X receptors (RXRs) are ligand-activated transcription factors that coordinate and regulate gene expression. This PPAR-RXR transcriptional complex plays a critical role in energy balance, including triglyceride metabolism, fatty acid handling and storage, and glucose homeostasis—processes in which dysregulation is characterized by obesity, diabetes, and atherosclerosis. PPARs and RXRs are also involved directly in inflammatory and vascular responses in endothelial and vascular smooth muscle cells [Plutzky J. Circ Res 2011].
The uncoupling of dose-response relations for different effects is the defining characteristic of a selective modulator. Thus, the pharmacological definition of a selective modulator is a ligand that, compared with a full agonist, differentially induces specific receptor effects [Higgins LS, DePaoli AM. Am J Clin Nutr 2010].
An ideal selective PPAR modulator (SPPARM) would be a potent and highly efficacious inducer of insulin sensitization, with low potency and/or low maximal activity for effects on adipose generation, loss of bone mineral density, fluid retention, and congestive heart failure. In such an ideal case, a therapeutic dose could be selected for a SPPARM at which antidiabetic benefits could be achieved while minimizing unwanted side effects [Higgins LS, Mantzoros CM. PPAR Res 2008].
Dual PPARα/PPARγ agonists have been designed to treat patients with insulin resistance and dyslipidemia. Known as glitazars, they combine the insulin-sensitizing effects of PPARγ activation with the lipid-lowering effects PPARα agonists, lowering triglycerides, increasing high-density lipoprotein cholesterol levels and insulin sensitivity, and reducing cardiovascular risk [Lalloyer F, Staels B. Arterioscler Thromb Vasc Biol 2010].
The SYNCHRONY study (NCT00388518), for example, aimed to establish the glucose-lowering and lipid-modifying effects and safety profile of the dual PPARα and PPARγ agonist aleglitazar. In a double-blind study, patients with T2DM (either drug-naive or pretreated with ≤two oral agents) were enrolled from 47 sites in 7 countries. After a single-blind, 4- to 5-week placebo run-in period, 332 patients were randomized (double-blind) via an interactive voice-response system to 16 weeks of treatment with aleglitazar at once-daily doses of 50 mug, 150 mug, 300 mug, or 600 mug; matching placebo (n=55 in each group); or open-label pioglitazone 45 mg once daily (n=57) as a reference.
The primary efficacy endpoint was the change in glycosylated HbA1C concentration from baseline to the end of treatment. Patients who received at least one dose of study drug and had at least one evaluable postbaseline HbA1C measurement were included in the efficacy analysis [Henry RR et al. Lancet 2009].
Findings showed that aleglitazar significantly reduced baseline HbA1C versus placebo in a dose-dependent manner, from −0.36% (95% CI, 0.00 to −0.70; p=0.048) with 50 mug to −1.35% (−0.99 to −1.70; p<0.0001) with 600 mug. The trend of changes over time suggested that the maximum effect of aleglitazar on HbA1C concentration was not yet reached after 16 weeks of treatment. Edema, hemodilution, and weight gain occurred in a dose-dependent manner. However, at aleglitazar doses less than 300 mug, no patients had congestive heart failure, frequency of edema was similar to placebo and less than with pioglitazone, and body weight gain was less than with pioglitazone (0.52 kg at 150 mug vs 1.06 kg) [Henry RR et al. Lancet 2009].
The favorable balance in the safety and efficacy profile of aleglitazar represents encouraging short-term clinical data for this agent and provides good evidence to enter Phase 3 investigation.
New insights into fundamental aspects of PPAR and RXR biology and their actions in the vasculature continue to be discovered. Although RXRs are obligate heterodimeric partners for PPAR action, the part that RXRs and their endogenous retinoid mediators exert in the vessel wall is less well understood. Biological insights into PPAR-RXRs may help inform interpretation of clinical trials with synthetic PPAR agonists and prospects for future PPAR therapeutics [Plutzky J. Circ Res 2011].
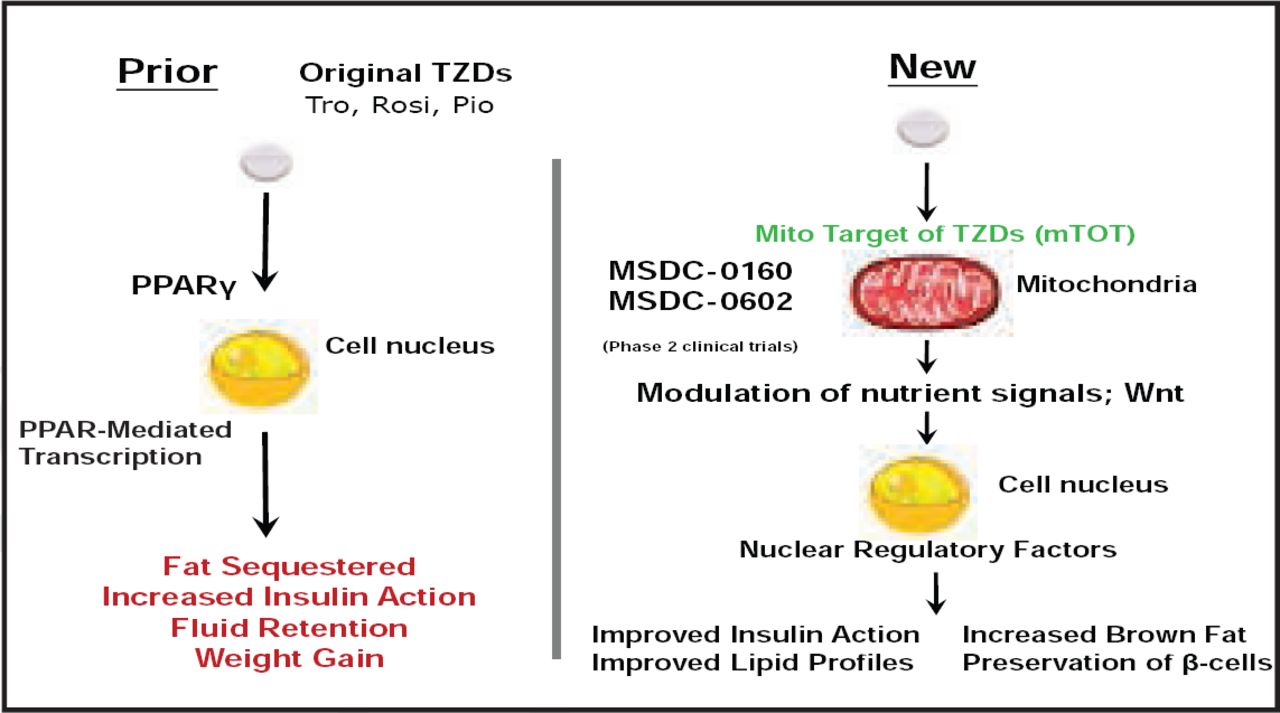
Novel antidiabetic drugs inhibit obesity-linked phosphorylation of PPARγ by CDK5 [Choi JH et al. Nature 2010]. Other new mechanisms of action for insulin sensitizers are under development (Figure 2). According to Dr. Plutzky, understanding of a novel non-PPAR mechanism of action presents a way forward.

Mechanism of Action for Insulin Sensitizers.
Reproduced with permission Metabolic Solutions Development Company LLC.
Next-Generation Metabolic Solutions
Charles Burant, MD, PhD, University of Michigan, Ann Arbor, Michigan, USA, started his presentation with a slide of the myriad of metabolic drugs in the pharmaceutical industry pipeline, ranging from a bile acid receptor agonist to a nicotinic acetylcholine receptor. He followed up with the 100+ targets that the new drugs are aimed at affecting and discussed novel compounds that are under development, such as elongase of long-chain fatty acids family 6 (ELOVL6).
As a rate-limiting enzyme for the elongation of saturated and monounsaturated long-chain fatty acids, ELOVL6 is abundantly expressed in lipogenic tissues, such as the liver, and its mRNA expression is upregulated in obese animal models. ELOVL6-deficient mice are protected from high-fat diet-induced insulin resistance, suggesting that ELOVL6 might be a new therapeutic target for diabetes [Shimamura K et al. Eur J Pharmacol 2010].
Another target—11 β-hydroxysteroid dehydrogenase type 1 (11 Beta HSD-1)—is an NADPH-dependent enzyme that is highly expressed in the liver, adipose tissue, and central nervous system [Hollis G, Huber R. Diabetes Obes Metab 2011]. The proposed mechanism of action is inhibition of the microsomal enzyme 11βHSD, which prevents the reduction of cortisone to the active hormone cortisol (or inhibition of corticosterone production in rodents). An orally active small molecule by Incyte Corporation, INCB13739 has completed a Phase 2a clinical trial, with promising outcomes [Rosenstock J et al. Diabetes Care 2010].
G-protein-coupled receptors are a family of approximately 850 proteins that have a wide variety of ligands with diverse effects on metabolism. The focus of research to date has been on fatty acid receptors. Fatty acid-binding GPCRs have effects on inflammation and insulin release and modulate insulin action. The proposed mechanism of the drug action is activation of GPR- enhanced insulin release in a glucose-dependent manner, leading to a decrease in inflammation that may improve insulin sensitivity [Im DS. Prostaglandins Other Lipid Mediat 2009].
Another target is the GLP-1 receptor. Activation of GLP-1 receptor signaling has pleiotropic effects, increasing insulin release and improving insulin action. ZY0G1 is an new, orally active, peptidomimetic molecular entity by Zydus Cadila. In vitro studies show potent activation of signaling in CJHO cells that express GLP-1R. In lean and obese mice, ZY0G1 improved glucose tolerance, decreased the area under the curve for insulin, and reduced weight gain and fasting glucose. In a Phase 1 human trial, ZY0G1 produced a dose-dependent pharmacokinetic response and a dose-dependent improvement in glucose tolerance in healthy human subjects [Peters AL. Clev Clin J Med 2009].
Activation of both GIP and GLP-1 receptors is expected to have synergistic effects on insulin secretion, increasing its secretion and biosynthesis as well as improving β-cell proliferation and survival [Chia CW et al. Diabetes 2009; Baggio LL, Drucker DJ. Gastroenterology 2007]. GIP/GLP-1 coagonists are peptide-engineered to bind and activate both the GIP and GLP-1 receptors [Tschop M, DiMarchi R et al. (in preparation)].
In animal models, the GIP/GLP-1 coagonist lowered glucose and weight in DIO mice in a dose-dependent manner, with no evidence of tachyphylaxis [Tschop M, DiMarchi R et al. (in preparation)]. The next-generation GIP/GLP-1 coagonist, an acylated coagonist with an extended half-life and high activity in vivo, has been tested in human studies since 2009.
In concluding his presentation, Dr. Burant cited alternative therapies as targets: weight loss, diet, exercise, and surgery. There are many targets for pharmacological therapy for diabetes, and many groups pursuing these targets. However, there is no perfect target and no perfect drug. “To expect one is unreasonable,” he concluded.
- © 2011 MD Conference Express
Tools









{kind=link}
{kind=link}
Table of contents
Cited By...
- No citing articles found.