

Summary
Activation of peroxisome proliferator-activated receptor (PPAR) is associated with dose-limiting side effects, such as edema and bone loss. This article describes recent efforts in the development of PPAR-sparing insulin sensitizers.
- endocrinology
- insulin
Activation of peroxisome proliferator-activated receptor (PPAR) is associated with dose-limiting side effects, such as edema and bone loss, said Jerry Colca, PhD, Metabolic Solutions Development Company, Kalamazoo, MI. In this presentation, Dr. Colca described recent efforts in the development of PPAR-sparing insulin sensitizers.
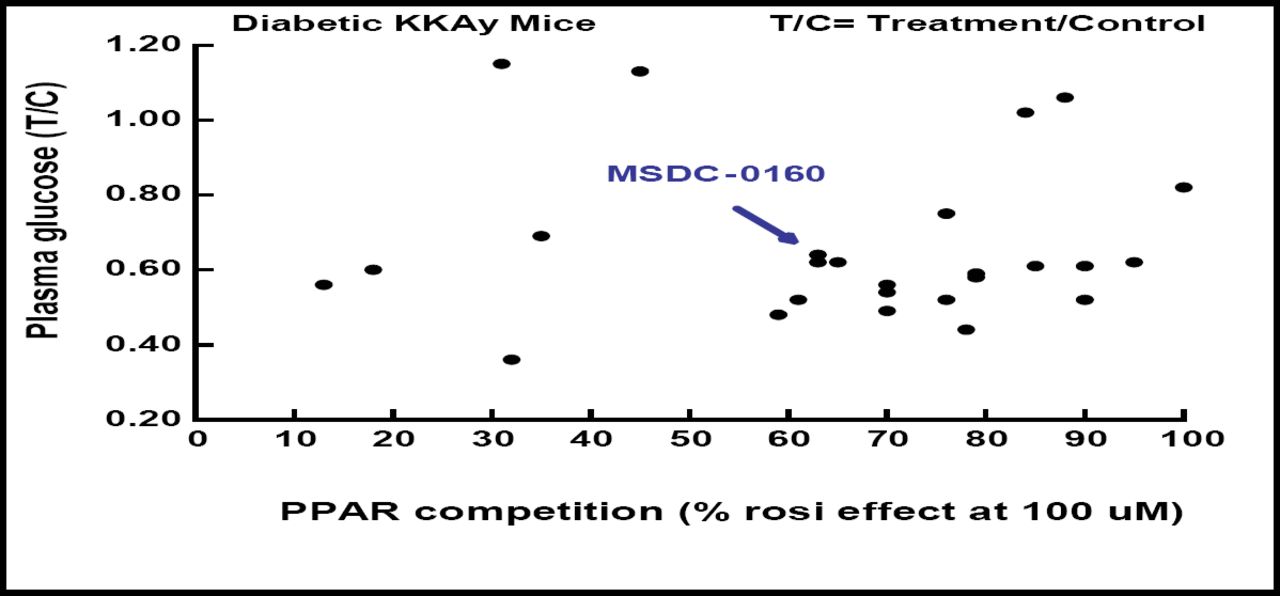
Dr. Colca and colleagues began their search for potent PPAR-sparing agents by testing more than 60 novel analogs for PPAR-binding activity using rosiglitazone as a positive control. Their work is creating analogs for which there is no correlation between PPAR–gamma binding and blood glucose lowering (Figure 1).

MSDC Analogs- No Correlation Between PPAR Binding and Pharmacology.
Candidate agents also have been screened for interaction with other PPAR subtypes, including PPAR-gamma, PPAR-alpha, and PPAR-delta. A prototypical candidate, MSDC-0160, also known as Mitoglitazone™, which showed little to no binding to any of the PPAR receptors, has been evaluated in phase 1 trials versus pioglitazone in normal human volunteers. The compound demonstrated one-a-day dosing kinetics and, like pioglitazone, increased circulating levels of the biomarker adiponectin.
Based on these findings, MSDC-0160 is under evaluation as a PPAR-sparing agent in phase 2 trials in a direct comparison with pioglitazone. In the phase 2 trials, Dr. Colca and colleagues also will evaluate whether PPAR-sparing insulin sensitizers, through direct interaction with mitochondria, provide more optimal insulin-sensitizing pharmacology in patients with type 2 diabetes.
Centrally Acting Bromocriptine Improves Glycemic Control
Researchers have observed that seasonal shifts from insulin resistance to insulin sensitivity were driven by fluctuations in hypothalamic neurotransmitter levels. Bromocriptine, a centrally acting dopamine D2 receptor agonist, acts by resetting the hypothalamic centers that regulate postprandial insulin-mediated glucose and lipid metabolism. When delivered in timed intervals, bromocriptine improves postprandial insulin responsiveness in insulin-resistant animal models.
Anthony H. Cincotta, PhD, Veroscience, Tiverton, RI, presented results from a study that compared bromocriptine with placebo among patients who were failing various peripherally acting oral hypoglycemic agents (OHAs). These patients were randomly assigned to additional treatment with bromocriptine (n=376) or placebo (n=183) for 52 weeks.
Baseline characteristics were similar in the two groups, including BMI (32.7 kg/m2 vs 32.3 kg/m2), fasting plasma glucose level (172.6 mg/dL vs 176.7 mg/dL), and HbA1c (8.3% vs 8.4%). The primary efficacy endpoint was change in glycemic control after 24 weeks. For the endpoint analysis, patients were classified according to background therapy: sulfonylurea ± any OHA; metformin ± any OHA; metformin plus sulfonylurea; or metformin plus glyburide.
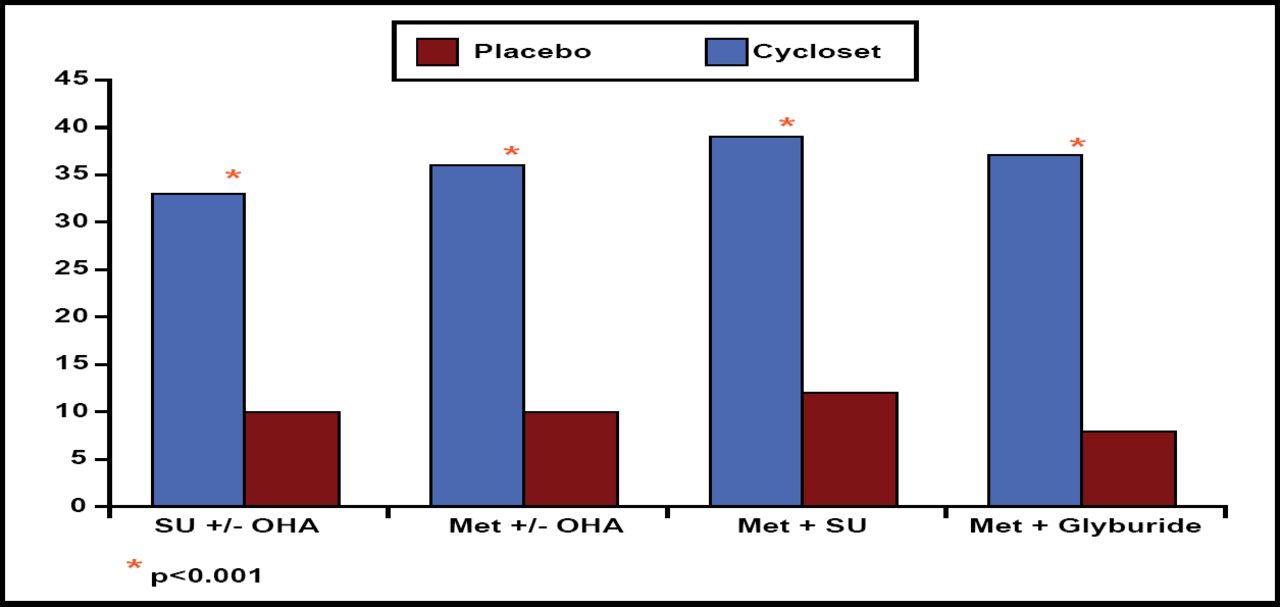
Daily morning pulsatile delivery of bromocriptine improved glycemic control after 24 weeks of therapy. Compared with placebo, bromocriptine significantly reduced HbA1c levels in all 4 patient subgroups (p<0.0003). In addition, treatment with bromocriptine allowed a significantly higher proportion of patients in each subgroup to achieve the target HbA1c goal of ≤7.0% at 24 weeks (p<0.001; Figure 2).

Percentage of Patients Reaching Goal HbA1c ≤7.0% After Week 24.
The safety analysis was performed after 52 weeks of therapy. The overall rate of serious adverse events was similar in the 2 treatment groups (HR=1.02). However, compared with placebo, bromocriptine was associated with a 42% reduction in the rate of adverse cardiovascular events, including myocardial infarction, stroke, heart failure hospitalization, angina, or revascularization surgery (HR=0.58; 95% CI 0.35, 0.96).
In summary, bromocriptine improves glycemic control and reduces the risk of cardiovascular events as adjunctive therapy in patients who are failing a range of OHAs. “Morning bromocriptine therapy for type 2 diabetes represents a potentially new approach in treating t he microvascular and macrovascular complications of this disease,” Dr. Cincotta concluded.
Teglicar, A Novel CPT1 Inhibitor, Reduces Fasting Blood Glucose
Teglicar is a reversible and selective inhibitor of carnitine palmitoyltransferase 1 (CPT1), a liver enzyme that is involved in postprandial gluconeogenesis. Giovanni Valentini, MD, Sigma-Tau, Pomezia, Italy, described preliminary findings of a phase 1 study that evaluated teglicar in patients with type 2 diabetes mellitus (T2DM).
The trial included 40 patients with T2DM that was controlled by diet alone or by hypoglycemic treatment with a single oral agent. Patients were randomly assigned to one of the following treatment groups: preprandial teglicar 150 mg (n=8); postprandial teglicar 150 mg (n=8); postprandial teglicar 450 mg (n=16); or placebo (n=8). Safety and efficacy were measured after an overnight fast at baseline (Day 1) and after the last day of treatment (Day 16).
The postprandial 450 mg dose of teglicar provided significant benefits. Compared with placebo, 450 mg teglicar significantly lowered mean fasting blood glucose (p=0.096). In addition, teglicar 450 mg reduced insulin resistance (p<0.04) and improved the homeostasis model assessment (HOMA) index compared with placebo.
In the safety analysis, teglicar was well tolerated and showed no clinically relevant changes in vital signs, electrocardiographic (ECG) findings, or laboratory parameters. In the 450 mg teglicar group, the most common adverse events were heartburn (n=4) and diarrhea (n=4).
“The statistically significant results obtained on reduction of tissue insulin resistance and a clear tendency to reduce fasting blood glucose make teglicar a promising tool in the control of fasting blood glucose in those patients where other glucose-lowering drugs fail to control nocturnal hepatic glucose production,” Prof. Valentini concluded.
Glucokinase Inhibition with RO4389620 Improves Fasting and Postprandial Glucose
Glucokinase (GK) is an enzyme that plays an important role in regulating glucose homeostasis. Jianguo Zhi, PhD, Hoffmann-La Roche Inc., Nutley, NJ, presented data on RO4389620, an investigational GK activator that has shown hypoglycemic effects in animal studies.
The phase 1 trial was designed to evaluate the safety and tolerability of RO4389620 after repeated dosing in 59 patients with T2DM. Patients were given RO4389620 or placebo as a single dose on Day 1, followed by multiple doses from Day 3 to Day 8. The dosing schedule included 10, 25, 50, 100, and 200 mg twice daily for 5.5 days or 200 mg once daily for 6 days.
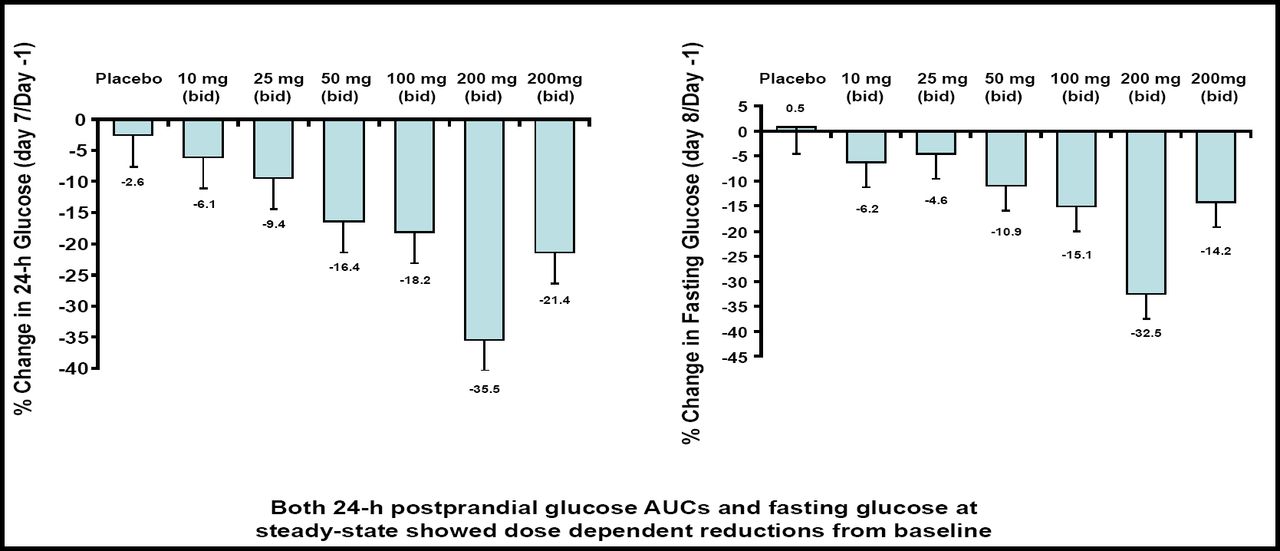
In the pharmacokinetic analysis, Dr. Zhi and colleagues found a linear correlation between dose and exposure, with no appreciable drug accumulation or food effect. In addition, RO4389620 was associated with dose-dependent reductions in 24-hour postprandial glucose areas under the curve (AUCs) and fasting glucose. At the 200 mg twice-daily dose, RO4389620 reduced the postprandial glucose AUC by 35.5% and fasting glucose by 32.5% compared with baseline (Figure 3). At this dose, Dr. Zhi also observed a mild increase in C-peptide (16%) and insulin secretion (10% to 19%).

RO4389620 Reduces Postprandial and Fasting Glucose.
RO4389620 was well tolerated, with no evidence of clinically significant laboratory, vital sign, or ECG abnormalities. Symptomatic hypoglycemia was the only dose-limiting adverse event, and no cases of severe hypoglycemia were reported.
In summary, RO4389620 demonstrated rapid, dose-dependent glucose lowering over 24 hours, with no safety concerns except mild to moderate hypoglycemia at the highest doses. Findings from this trial demonstrate the potential utility of RO4389620 in the treatment of T2DM, Dr. Zhi said.
- © 2008 MD Conference Express
Tools









{kind=link}
{kind=link}
{kind=link}
Table of contents
Cited By...
- No citing articles found.