

Summary
Although combination therapies can help fight therapy resistance when using personalized cancer treatment, the seemingly endless number of possible combinations necessitates strategies to identify rational drug combinations, as discussed in this article.
- Soft Tissue Cancers
- Gastrointestinal Cancers
- Oncology Genomics
- Soft Tissue Cancers
- Gastrointestinal Cancers
- Oncology
- Oncology Genomics
Although combination therapies can help fight therapy resistance when using personalized cancer treatment, the seemingly endless number of possible combinations necessitates strategies to identify rational drug combinations, said René Bernards, PhD, The Netherlands Cancer Institute, Amsterdam, The Netherlands.
Many of the new cancer drugs elicit powerful initial responses that lead to dramatic effects on progression-free survival (PFS) [Chapman PB et al. N Engl J Med. 2011; Maemondo M et al. N Engl J Med. 2010], but single-agent therapy is of limited benefit with the development of resistance. This resistance leads to convergence of the PFS curves between targeted therapies and standard chemotherapy and prevents significant gains in overall survival. The solution to resistance is combination therapy, said Prof Bernards. But with nearly 1000 cancer drugs in development, testing each 2-way combination in just 1000 patients would require nearly half a billion subjects. Preclinical models are therefore needed to help select the most effective combinations of drugs up front before they are tested in patients.
The difficulty in choosing relevant targets is exemplified by the differential response to BRAF inhibition in 2 forms of BRAF-mutant cancers: melanoma and colon cancer [Flaherty KT et al. N Engl J Med. 2010]. Melanoma is highly responsive to BRAF inhibition, but colon cancer is not, demonstrating that the context matters in which a mutation is present. In colon cancer, a secondary pathway activation may preclude BRAF inhibition from being effective. Most likely, the moment a pathway is inhibited that is signaling from the cell surface to the nucleus, a secondary pathway becomes activated that maintains cell viability, explained Prof Bernards. Often, these secondary pathways are unknown; learning them would enable rational combination therapy.
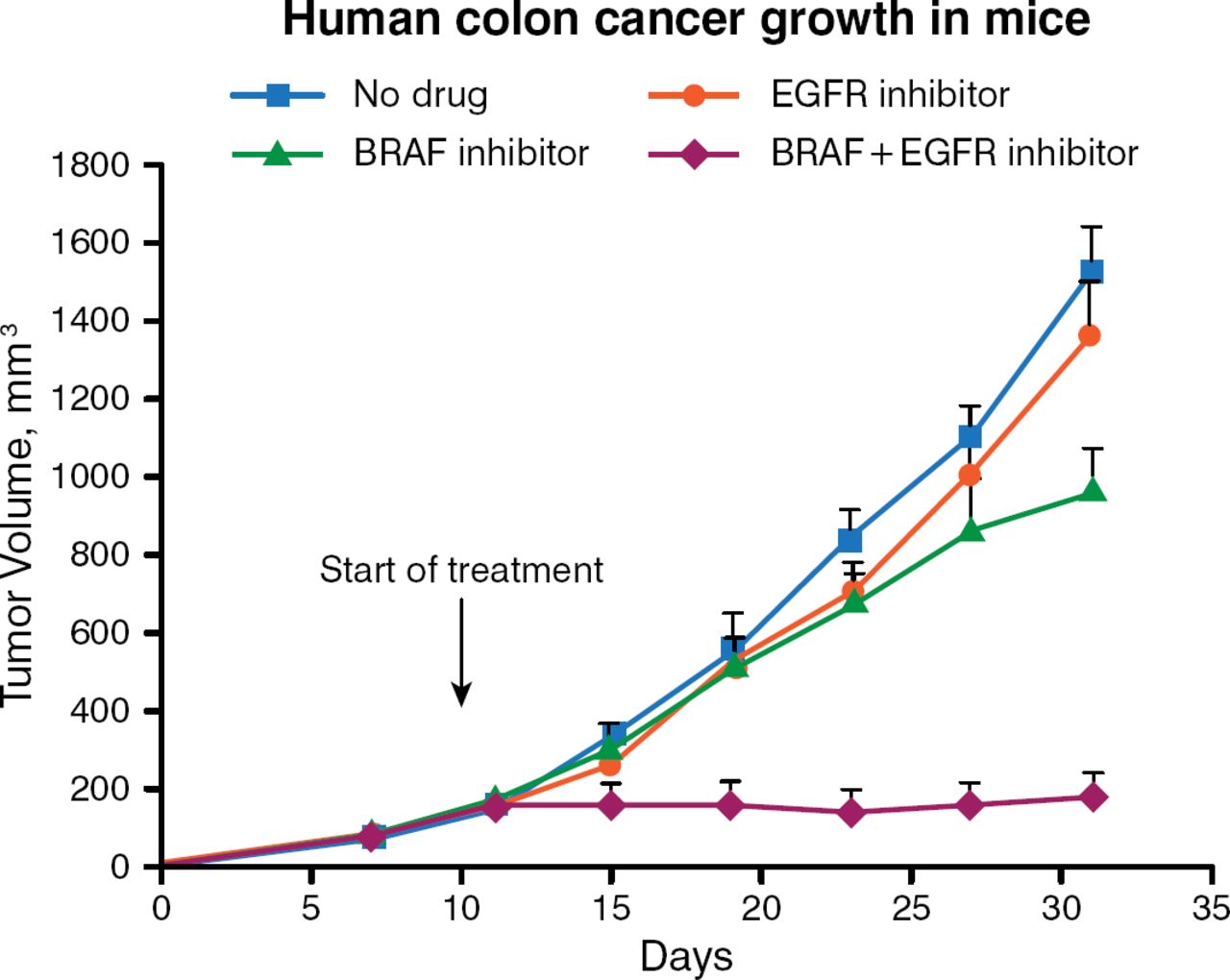
In the laboratory, BRAFV600E mutant colorectal cancer cell lines also are found to be less responsive to increasing concentrations of PLX4032 (vemurafenib) than are melanomas having the same mutation. Functional genetic screens are being used to find powerful combinations of cancer drugs by exploiting the concept of “synthetic lethality,” or simultaneous inactivation of 2 pathways leading to cell death, he said. Using RNA interference-based genetic screens with collections of small hairpin RNAs that target gene families for which drugs have been developed, his laboratory searches for genes whose inactivation is particularly synergistic with anticancer drugs. Using this screening approach, the gene that encodes EGFR was found to be a rational target. Inhibiting BRAF causes feedback activation of EGFR, leading to activation of AKT (protein kinase B) signaling. Inhibiting EGFR makes BRAF-mutant colorectal cancer cells vulnerable to BRAF inhibition. Combining a BRAF inhibitor with an EGFR inhibitor silences both the EGFR and AKT pathways, suppressing BRAF-mutant colon cancer cell growth (Figure 1) [Prahallad A et al. Nature. 2012].

Suppression of Colon Cancer Growth by Simultaneously Inhibiting EGFR and BRAF
Reprinted by permission from Macmillan Publishers Ltd: Nature. Prahallad A et al. Unresponsiveness of colon cancer to BRAF(V600E) inhibition through feedback activation of EGFR. 2012;483:100–104. © 2012.
Similarly, in KRAS-mutant tumors, single-agent MEK inhibition in lung and colon cancers is not effective. Adding an EGFR inhibitor is not synergistic. Apparently, even though the BRAF and KRAS pathways are mutually exclusive, the wiring of KRAS-mutant cells is different from that of BRAF-mutant cells, said Prof Bernards. A synthetic lethality screen for enhancers of MEK inhibitors reveals that suppression of ERBB3 receptor tyrosine kinase sensitizes KRAS-mutant cancers to MEK inhibitors [Sun C et al. Cell Rep. 2014]. Drugs that target both EGFR and ERBB2, each of which is capable of forming heterodimers with ERBB3, have been shown to sensitize KRAS-mutant cells to a MEK inhibitor. Dual inhibitors of EGFR and ERBB2 are afatinib and dacomitinib, and indeed these agents demonstrate strong synergy with a MEK inhibitor, he said.
The potential for synergy with a BRAF inhibitor by inhibiting a phosphatase in BRAF-mutant colon cancer is being explored. One such kinase that shows synergy is PTPN11, also known as SHP2. It is a member of a family of cytoplasmic Src 2 homology 2 (SH2) domain-containing protein tyrosine phosphatases. The N-SH2 domain selectively binds to phosphotyrosyl motifs on receptor tyrosine kinases (RTKs), and it is required for activation of RAS signaling downstream of RTKs. BRAF-mutant colorectal cancer cells that lack PTPN11 are sensitive to vemurafenib. Knocking out PTPN11 also confers sensitivity to MEK inhibition in KRAS-mutant tumors.
In conclusion, the data show that many feedback systems are in play to reactivate cell surface receptors when a single pathway is inhibited. EGFR, KIT, c-MET receptors, and ERBB3 use PTPN11 for signaling downstream, and removing PTPN11 can alter feedback signaling. Inhibition of PTPN11, therefore, is a promising target for treating any cancer that suffers from RTK reactivation after primary therapy.
- © 2014 MD Conference Express®
Tools









{kind=link}
Table of contents
Cited By...
- No citing articles found.