

Summary
Tumor genomic profiling in clinical studies and the integration of tumor genomics into clinical practice are rapidly becoming the standard of care. This article discusses methods to apply genomics to guide clinical investigation, next-generation sequencing, how homologous recombination acts as a target and as a predictive and prognostic biomarker, and the therapeutic dissection of lung cancer.
- Oncology Genomics
- Oncology Genomics
- Oncology
Tumor genomic profiling in clinical studies and the integration of tumor genomics into clinical practice are rapidly becoming the standard of care.
Paul Workman, PhD, DSc, Institute of Cancer Research, London, United Kingdom, spoke about applying genomics to guide clinical investigation, noting that an improved understanding of the genetics and physiology of cancers will drive new treatments.
Priority must be given to extending the “druggable” cancer genome to provide drugs for all key pathways and nodes; currently only about 5% of the cancer genome is drugged, and the remainder urgently needs to be completed [Patel MN et al. Nat Rev Drug Discov 2013]. To this end, a computational knowledge base resource for cancer translational research and drug discovery, canSAR, has been developed. It allows the integration of gene sequencing data and the prioritization of targets for drug discovery as the data emerge. Using computational techniques, targets can be validated more quickly from genome sequencing.
A network understanding of cancer is currently lacking. Historical drug discovery has been focused on limited networks, said Prof. Workman, but druggable targets in networks outside of previous bias have been identified. His group has proposed that combinatorial targeting of different subnetworks may reduce drug resistance.
Heat shock protein 90 (HSP90) inhibitors may turn out to be the first powerful network-based drugs, he said. HSP90 is a molecular chaperone that “talks” to a large number of proteins in the cell, particularly oncogene products and mutated, amplified, and overexpressed genes. It is important for folding, function, activation, and stability of many oncogenic client proteins. It is especially important for mutant or amplified client oncoproteins and drug-resistant forms (eg, ERBB2, EGFR, ALK, BRAF, AR). HSP90 inhibitors hit multiple cancer vulnerabilities, such as targets, pathways, and hallmark traits, and thereby show tumor selectivity. They may act as the first cancer network drugs with the potential to overcome and prevent drug resistance, said Dr. Workman.
BRAF-mutant colorectal cancers are predicted to be responsive to HSP90 inhibitors, as HSP90 inhibition has been shown to block signaling output from mutant BRAF but not wild-type BRAF. This blockade of signaling is related to chaperone deprivation by client proteins and HSP90 itself. Driving this depletion is CUL5, a gene that is required for degradation of multiple kinase clients [Samant RS et al. Proc Natl Acad Sci U S A 2014], said Prof. Workman. CUL5 knockdown delays HSP90-induced signaling disruption of client proteins, indicating a key role. CUL5 could therefore be a potential biomarker of sensitivity and of a resistance mechanism.
Elaine Mardis, PhD, Washington University, St. Louis, Missouri, USA, provided the basics on next-generation sequencing and on how “massively parallel sequencing” works. Massively parallel sequencing is a high-throughput deoxyribonucleic acid (DNA) sequencing technology that is capable of sequencing large numbers of different DNA sequences in a single reaction.
All next-generation sequencing platforms require a library obtained by either amplification or ligation with custom linkers (adapters). Each library fragment is amplified on a solid surface with covalently attached synthetic adapters that hybridize the library adapters.
Although polymerase chain reaction (PCR) is an effective vehicle for amplifying DNA, in the library construction, it can introduce preferential amplification (referred to as “jackpotting”) of certain fragments and the need to “de-duplicate” identical copies (duplicate reads) that can occur. Low-input DNA amounts favor jackpotting due to lack of complexity in the library. PCR can also introduce false-positive artifacts due to substitution errors by the polymerase, which is more problematic if it occurs in early PCR cycles because the error appears as a true variant. Cluster formation is also a type of PCR, which can also introduce bias in amplifying high and low G+C content in the fragment, and as a result, coverage is reduced at these loci, potentially leading to false negatives.
Hybrid capture improves transcriptome analysis on low-input and archived samples. In hybrid capture, fragments from a whole genome library are selected by combing with probes that correspond to most human exons or gene targets. The probe DNAs are biotinylated, making selection from solution with streptavidin magnetic beads an effective means of purification.
Next-generation sequencing is unique in that direct step-by-step detection is performed of each nucleotide base that is being incorporated onto each amplified library fragment being sequenced. Next-generation sequencing is typically referred to as massively parallel sequencing because the stepwise process involves hundreds of thousands to hundreds of millions of reactions that are detected per instrument run.
Because each library fragment starts as a single piece of DNA, the amount of DNA that is contributed by that particular site in the genome and the particular ribonucleic acid in the transcriptome is measured quantitatively. The digital reads, if appropriately treated, enable direct quantitative comparisons between samples. They are typically shorter read lengths than are capillary sequencers, which deliver hundreds of nucleotides.
Christine S. Walsh, MD, Cedars-Sinai Medical Center, Los Angeles, California, USA, reviewed how homologous recombination (HR) acts as a target and as a predictive and prognostic biomarker.
Over the past 20 years, HR has become a target for cancer treatment, starting at the time BRCA1 and BRCA2 were isolated. HR is an error-free DNA repair pathway for double-strand DNA breaks, Dr. Walsh explained. BRCA1 and BRCA2 are genome caretaker genes that participate in the HR pathway.
Ovarian cancers can be deficient in HR either through deficiency of BRCA1 or BRCA2 through germline or somatic mutations or epigenetic BRCA1 silencing. Other HR defects may have a “BRCAness” phenotype in which they act like BRCA-mutated tumors without a germline or somatic BRCA inactivation [Cancer Genome Atlas Research Network Nature 2011].
BRCA1/2-positive ovarian cancer patients have better survival than negative patients [Yang D et al. JAMA 2011; Chetrit A et al. J Clin Oncol 2008; Cass I et al. Cancer 2003], an observation that extends to other HR-deficient cancers. HR deficiency in ovarian cancer predicts platinum sensitivity and better survival [Pennington KP et al. Clin Cancer Res 2014]. BRCA1/2-positive breast and ovarian cancers also have an enhanced response to doxorubicin/pegylated liposomal doxorubicin [Graeser M et al. Clin Cancer Res 2010; Safra T et al. Mol Cancer Ther 2011].
HR deficiency correlates with selective response to poly–adenosine diphosphate ribose polymerase (PARP) inhibitors [Fong PC et al. N Engl J Med 2009; McCabe N et al. Cancer Res 2006]. PARP is a family of enzymes involved in base excision repair, which is the key pathway in the repair of single-strand DNA breaks. BRCA-deficient cells are 1000 times more sensitive to PARP inhibition than wild-type cells [Bryant HE et al. Nature 2005; Farmer H et al. Nature 2005], and this observation led to the concept of synthetic lethality, in which a deficiency of both PARP and BRCA has a lethal effect.
PARP inhibitors are thought to work through direct blockade of PARP enzymatic activity and PARP accumulation on DNA in a process called PARP trapping. A number of PARP inhibitors are in clinical development. Olaparib was associated with responses limited to BRCA carriers with ovarian cancer in Phase 1 study [Audeh MW et al. Lancet 2010; Fong PC et al. J Clin Oncol 2010; Fong PC et al. N Engl J Med 2009; Tutt A et al. Lancet 2010] and some responses in sporadic ovarian cancer in Phase 2 [Gelmon KA et al. Lancet Oncol 2011]. These responses correlated with platinum sensitivity. In a Phase 2 study, olaparib produced a higher overall response rate and a better toxicity profile compared with standard chemotherapy but no difference in progression-free survival in patients with BRCA1-positive or BRCA2-positive platinum-resistant ovarian cancer [Kaye SB et al. J Clin Oncol 2012].
In the maintenance setting in ovarian cancer, olaparib was associated with greater progression-free survival but no difference in overall survival compared with placebo, leading to an arrest in its development [Ledermann J et al. N Engl J Med 2012]. A post hoc analysis showed that in the BRCA carriers, however, progression-free survival was 11.2 months with olaparib versus 4.1 months with placebo (p<0.01) [Ledermann J et al. J Clin Oncol 2013], after which the manufacturer resumed development. Phase 3 trials as maintenance therapy have been launched. Other PARP inhibitors that have shown activity in advanced solid tumors are veliparib, rucaparib, and niraparib.
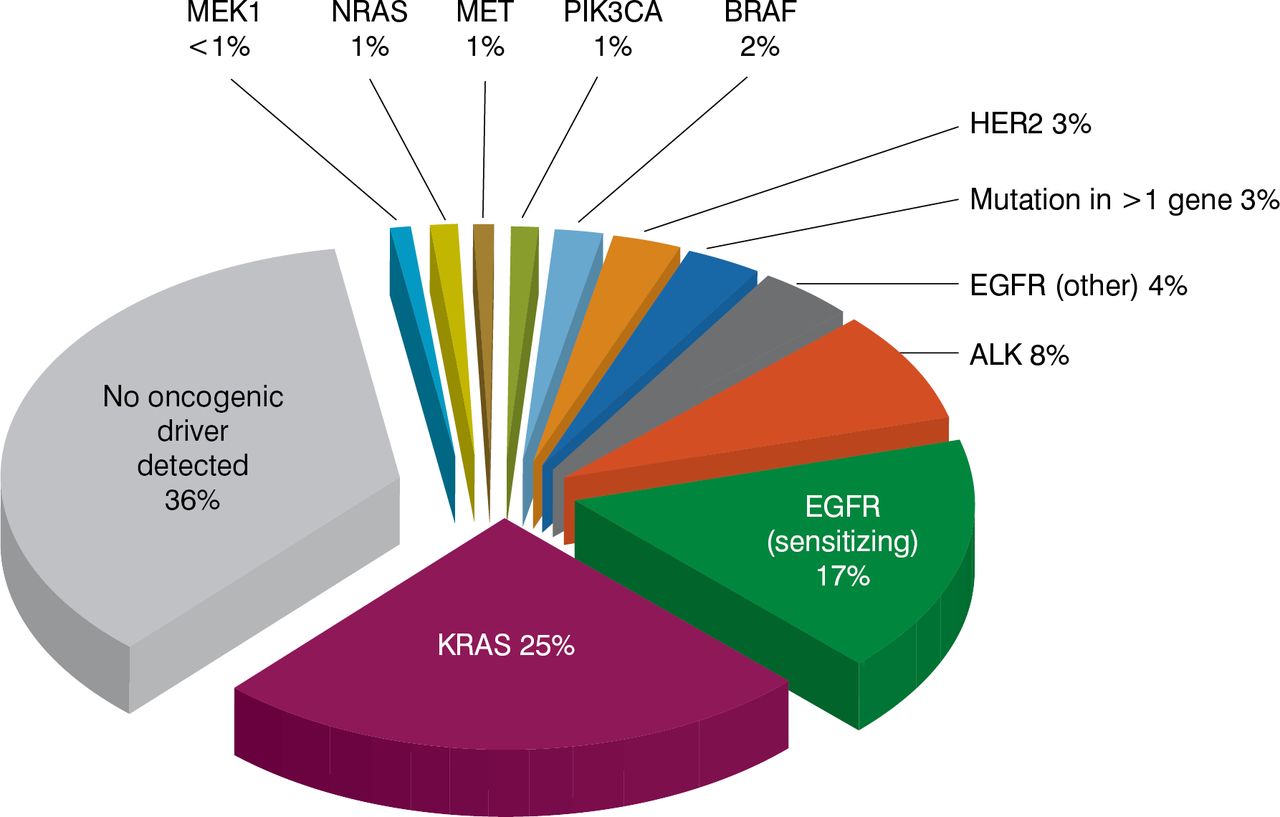
Charles M. Rudin, MD, PhD, Memorial Sloan Kettering Cancer Center, New York, New York, USA, spoke on therapeutic dissection of lung cancer, which has an exceptionally high mutation burden, especially in tobacco smokers. The largest slice of the lung cancer pie is adenocarcinoma. Genomic analysis to identify at least the large slices of the lung adenocarcinoma pie is a standard of care, as driver mutations have a clear clinical significance, he said. The most frequent mutation observed is KRAS (25%), followed by EGFRsensitizing mutations (17%), ALK translocations (8%), EGFR-non-sensitizing mutations (4%), and others [Kris MG et al. JAMA 2014]. These segments are essentially nonoverlapping, representing distinct subsets with distinct biology and distinct targetable driver alterations. No oncogenic driver is found in about one-third of lung adenocarcinomas. Survival probability is higher when lung adenocarcinoma has a driver mutation for which a targeted therapy exists compared with one for which no targeted agent exists or one that has no driver identified [Kris MG et al. JAMA 2014].

Detection of Driver Mutations in Lung Adenocarcinoma
Similar analyses of squamous cell lung cancer to guide clinical trial enrollment has begun. The most commonly altered gene is TP53, followed by CDKN2A, PTEN, PIK3CA, and a variety of others [Cancer Genome Atlas Research Network. Nature 2012]. Relevant mutational subsets of squamous cell carcinoma are PTEN loss (22%), FGFR1 amplification, PIK3CA mutation, and others, with one-third having no identifiable driver alteration [Drilon A et al. Lancet Oncol 2012].
Potential drivers of small-cell lung cancer have recently been identified, with a predominance of the tumor suppressors TP53 and RB1 rather than oncogenes [Peifer M et al. Nat Genet 2012]. These data are emerging, and more work is needed to make these therapeutically intractable.
- © 2014 MD Conference Express®
Tools









{kind=link}
Table of contents
Cited By...
- No citing articles found.