

Summary
The oral direct factor Xa inhibitor betrixaban, at doses of 40 mg, 60 mg, and 80 mg once daily, is safe and well tolerated compared with dose-adjusted warfarin in patients with nonvalvular atrial fibrillation (AF) or atrial flutter. This article presents results from the Phase II, randomized, multicenter EXPLORE-Xa Trial [NCT00742859].
- arrhythmias clinical trials
The oral direct factor Xa inhibitor betrixaban, at doses of 40 mg, 60 mg, and 80 mg once daily, is safe and well tolerated compared with dose-adjusted warfarin in patients with nonvalvular atrial fibrillation (AF) or atrial flutter. Michael D. Ezekowitz, MD, PhD, Vice President, Lankenau Institute for Medical Research, Thomas Jefferson Medical College, Wynnewood, PA, presented results from the Phase II, randomized, multicenter EXPLORE-Xa Trial (NCT00742859).
Dr. Ezekowitz pointed out a few important characteristics of betrixaban, including its effective half-life of approximately 20 hours and the fact that it is being codeveloped with an antidote. No dose adjustments for renal impairment or major drug interactions were anticipated during this trial, because betrixaban is excreted mostly unchanged through bile, with minimal renal excretion, and it is not a substrate for the CYP450 system.
In EXPLORE-Xa, 508 patients with nonvalvular AF and at least one risk factor for stroke were randomized to receive betrixaban 40 mg (n=127), 60 mg (n=127), or 80 mg (n=127) or open-label warfarin (n=127) with an international normalized ratio goal of 2 to 3. The mean age was 74 years, and the median follow-up was 4.9 months (minimum follow-up 3 months; maximum follow-up 12 months). Patients were excluded from participation in the study if they had active endocarditis, AF due to reversible cases or mechanical heart valve, scheduled major surgery or pulmonary vein ablation, or repeated systolic blood pressure >160 mmHg; had received hemodialysis within one year; or experienced a recent ischemic stroke, systemic embolic event, or acute coronary syndrome within 30 days. The primary endpoint was occurrence of major or clinically relevant nonmajor bleeding. The secondary endpoints were time to occurrence of any bleeding (major, clinically relevant nonmajor, and minimal) and time to occurrence of death, stroke, myocardial infarction (MI), or other systemic embolism.
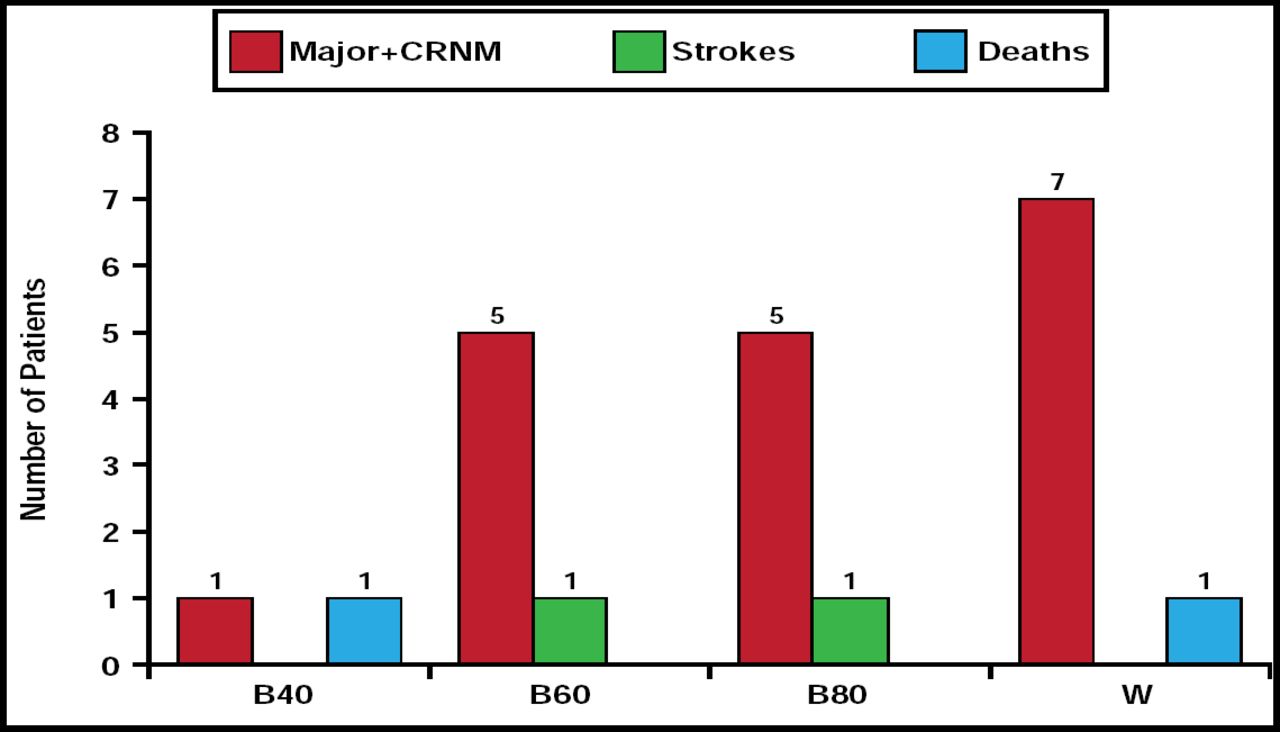
At 3 months, the rate of major or clinically relevant nonmajor bleeding in the betrixaban 40-mg group (n=1) was significantly less than in the warfarin group (n=4). Bleeding rates in the groups that received betrixaban 60 mg (n=4) and 80 mg (n=5) were comparable with rates that were observed in the warfarin group. The number of strokes and deaths was low in all treatment groups (Figure 1). Patients who received the 40-mg betrixaban dose demonstrated a slight increase in d-dimer from baseline, and there was a trend toward a dose response with d-dimer activity across the dose spectrum.

Bleeds, Strokes, and Deaths.
Reproduced with permission from M. Ezekowitz., MD, PhD.
Adverse events were equally distributed among the groups, with the exception of gastrointestinal adverse events. The incidence of vomiting, nausea, and diarrhea was more common in patients who received betrixaban. There was no difference in the incidence of alanine aminotransferase >2x the upper limit of normal in any of the groups (2.4% for betrixaban and warfarin groups).
Betrixaban 40 mg, 60 mg, and 80 mg appear to be well tolerated in patients with AF or atrial flutter. There was a dose-dependent effect on the primary endpoint of major or clinically relevant nonmajor bleeding that was associated with betrixaban therapy. More comprehensive evaluation in a larger study population is needed to determine the safety and efficacy of betrixaban therapy.
- © 2010 MD Conference Express
Tools









{kind=link}
Table of contents
Cited By...
- No citing articles found.